

Predicting functional gene links from phylogenetic-statistical analyses of whole genomes
- PMID: 16103904
- PMCID: PMC1183509
- DOI: 10.1371/journal.pcbi.0010003
Predicting functional gene links from phylogenetic-statistical analyses of whole genomes
Abstract
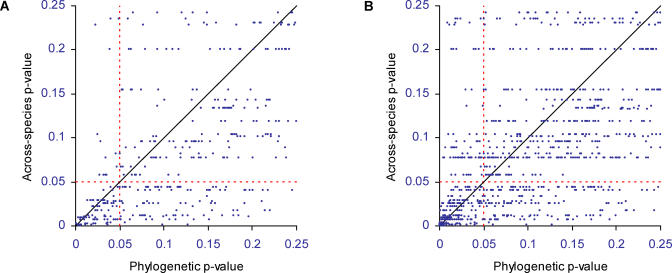
An important element of the developing field of proteomics is to understand protein-protein interactions and other functional links amongst genes. Across-species correlation methods for detecting functional links work on the premise that functionally linked proteins will tend to show a common pattern of presence and absence across a range of genomes. We describe a maximum likelihood statistical model for predicting functional gene linkages. The method detects independent instances of the correlated gain or loss of pairs of proteins on phylogenetic trees, reducing the high rates of false positives observed in conventional across-species methods that do not explicitly incorporate a phylogeny. We show, in a dataset of 10,551 protein pairs, that the phylogenetic method improves by up to 35% on across-species analyses at identifying known functionally linked proteins. The method shows that protein pairs with at least two to three correlated events of gain or loss are almost certainly functionally linked. Contingent evolution, in which one gene's presence or absence depends upon the presence of another, can also be detected phylogenetically, and may identify genes whose functional significance depends upon its interaction with other genes. Incorporating phylogenetic information improves the prediction of functional linkages. The improvement derives from having a lower rate of false positives and from detecting trends that across-species analyses miss. Phylogenetic methods can easily be incorporated into the screening of large-scale bioinformatics datasets to identify sets of protein links and to characterise gene networks.
Conflict of interest statement
Figures
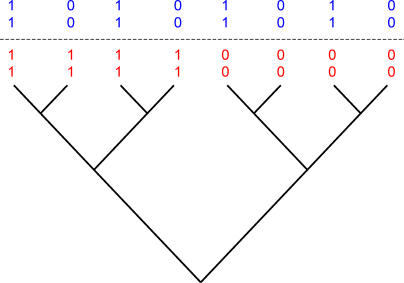
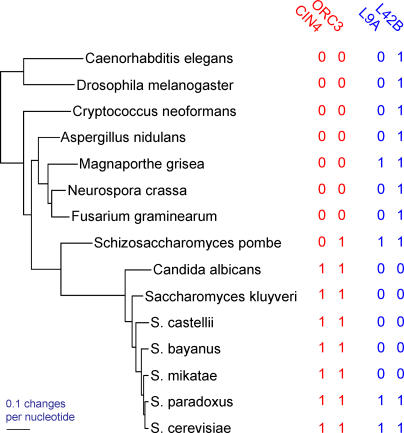
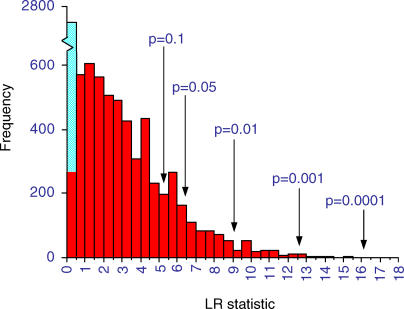
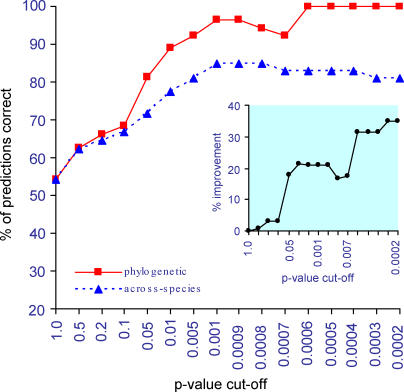
References
-
- Eisenberg D, Marcotte EM, Xenarios I, Yeates TO. Protein function in the post-genomic era. Nature. 2000;405:823–826. - PubMed
-
- Date SV, Marcotte EM. Discovery of uncharacterized cellular systems by genome-wide analysis of functional linkages. Nat Biotechnol. 2003;21:1055–1062. - PubMed
-
- Pazos F, Valencia A. Similarity of phylogenetic trees as indicator of protein-protein interaction. Protein Eng. 2001;14:609–614. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
