

Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface
- PMID: 16107719
- PMCID: PMC1190273
- DOI: 10.1128/MCB.25.17.7734-7742.2005
Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface
Abstract
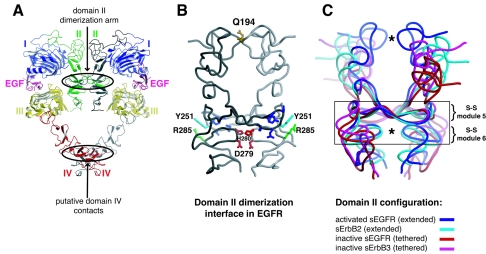
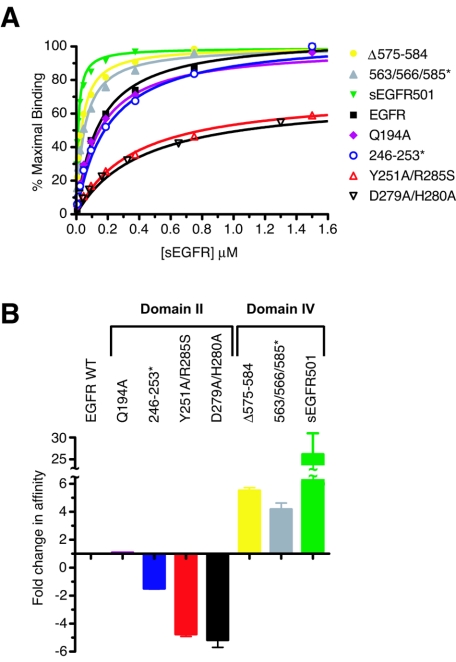
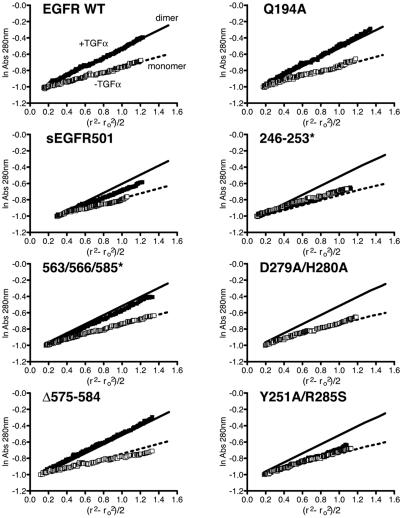
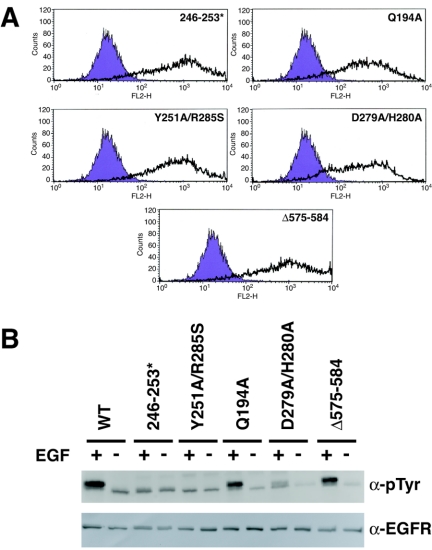
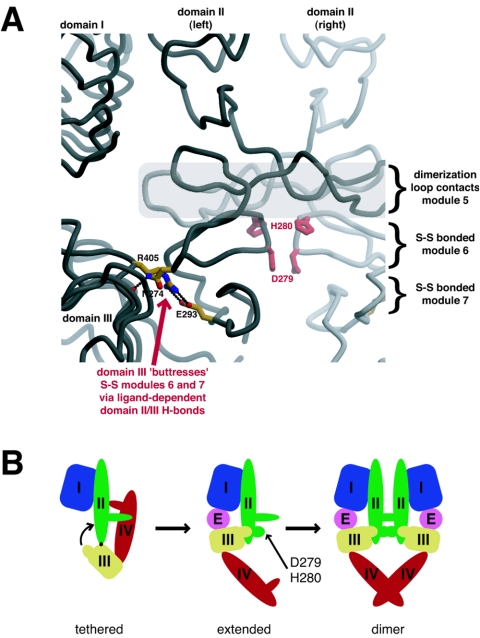
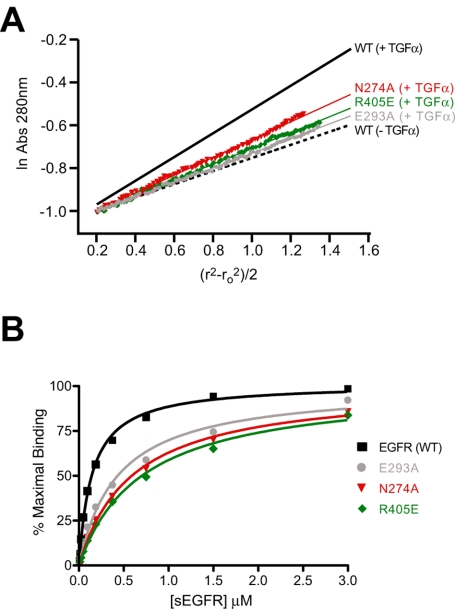
Structural studies have shown that ligand-induced epidermal growth factor receptor (EGFR) dimerization involves major domain rearrangements that expose a critical dimerization arm. However, simply exposing this arm is not sufficient for receptor dimerization, suggesting that additional ligand-induced dimer contacts are required. To map these contributions to the dimer interface, we individually mutated each contact suggested by crystallographic studies and analyzed the effects on receptor dimerization, activation, and ligand binding. We find that domain II contributes >90% of the driving energy for dimerization of the extracellular region, with domain IV adding little. Within domain II, the dimerization arm forms much of the dimer interface, as expected. However, a loop from the sixth disulfide-bonded module (immediately C-terminal to the dimerization arm) also makes a critical contribution. Specific ligand-induced conformational changes in domain II are required for this loop to contribute to receptor dimerization, and we identify a set of ligand-induced intramolecular interactions that appear to be important in driving these changes, effectively "buttressing" the dimer interface. Our data also suggest that similar conformational changes may determine the specificity of ErbB receptor homo- versus heterodimerization.
Figures
References
-
- Arteaga, C. L. 2003. ErbB-targeted therapeutic approaches in human cancer. Exp. Cell Res. 284:122-130. - PubMed
-
- Beerli, R. R., and N. E. Hynes. 1996. Epidermal growth factor-related peptides activate distinct subsets of ErbB receptors and differ in their biological activities. J. Biol. Chem. 271:6071-6076. - PubMed
-
- Berezov, A., J. Chen, Q. Liu, H. T. Zhang, M. I. Greene, and R. Murali. 2002. Disabling receptor ensembles with rationally designed interface peptidomimetics. J. Biol. Chem. 277:28330-28339. - PubMed
-
- Berger, M. B., J. M. Mendrola, and M. A. Lemmon. 2004. ErbB3/HER3 does not homodimerize upon neuregulin binding at the cell surface. FEBS Lett. 569:332-336. - PubMed
-
- Blume-Jensen, P., and T. Hunter. 2001. Oncogenic kinase signaling. Nature 411:355-365. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous