

Dissecting the genetic complexity of human 6p deletion syndromes by using a region-specific, phenotype-driven mouse screen
- PMID: 16109771
- PMCID: PMC1194901
- DOI: 10.1073/pnas.0500584102
Dissecting the genetic complexity of human 6p deletion syndromes by using a region-specific, phenotype-driven mouse screen
Abstract
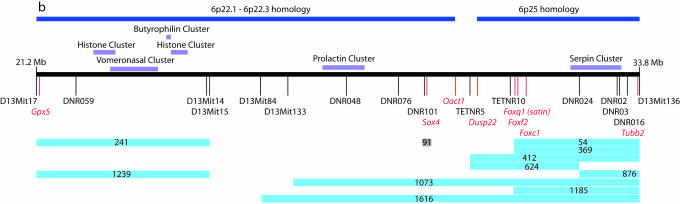
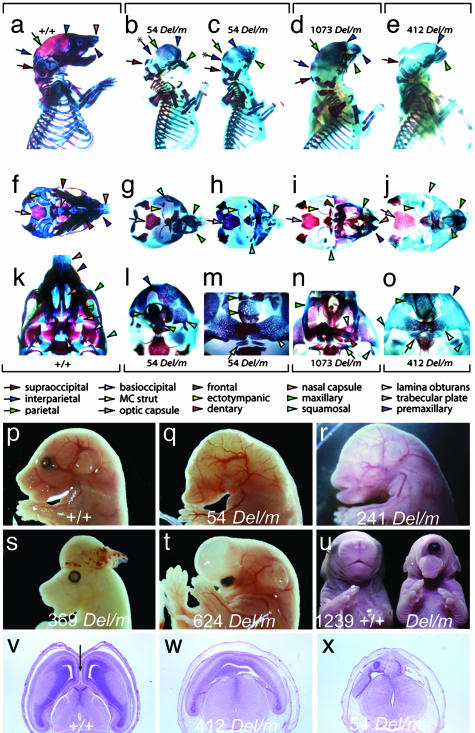
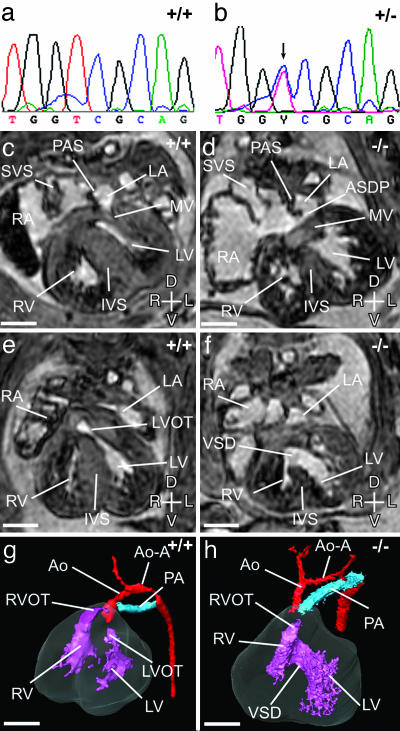
Monosomy of the human chromosome 6p terminal region results in a variety of congenital malformations that include brain, craniofacial, and organogenesis abnormalities. To examine the genetic basis of these phenotypes, we have carried out an unbiased functional analysis of the syntenic region of the mouse genome (proximal Mmu13). A genetic screen for recessive mutations in this region recovered thirteen lines with phenotypes relevant to a variety of clinical conditions. These include two loci that cause holoprosencephaly, two that underlie anophthalmia, one of which also contributes to other craniofacial abnormalities such as microcephaly, agnathia, and palatogenesis defects, and one locus responsible for developmental heart and kidney defects. Analysis of heterozygous carriers of these mutations shows that a high proportion of these loci manifest with behavioral activity and sensorimotor deficits in the heterozygous state. This finding argues for the systematic, reciprocal phenotypic assessment of dominant and recessive mouse mutants. In addition to providing a resource of single gene mutants that model 6p-associated disorders, the work reveals unsuspected genetic complexity at this region. In particular, many of the phenotypes associated with 6p deletions can be elicited by mutation in one of a number of genes. This finding implies that phenotypes associated with contiguous gene deletion syndromes can result not only from dosage sensitivity of one gene in the region but also from the combined effect of monosomy for multiple genes that function within the same biological process.
Figures
References
-
- Palmer, C. G., Bader, P., Slovak, M. L., Comings, D. E. & Pettenati, M. J. (1991) Am. J. Med. Genet. 39, 155-160. - PubMed
-
- Davies, A. F., Mirza, G., Sekhon, G., Turnpenny, P., Leroy, F., Speleman, F., Law, C., van Regemorter, N., Vamos, E., Flinter, F., et al. (1999) Hum. Genet. 104, 64-72. - PubMed
-
- Mirza, G., Williams, R. R., Mohammed, S., Clark, R., Newbury-Ecob, R., Baldinger, S., Flinter, F. & Ragoussis, J. (2004) Eur. J. Hum. Genet. 12, 718-728. - PubMed
-
- Hong, H. K., Lass, J. H. & Chakravarti, A. (1999) Hum. Mol. Genet. 8, 625-637. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
