

Tandem machine learning for the identification of genes regulated by transcription factors
- PMID: 16115317
- PMCID: PMC1208855
- DOI: 10.1186/1471-2105-6-204
Tandem machine learning for the identification of genes regulated by transcription factors
Abstract
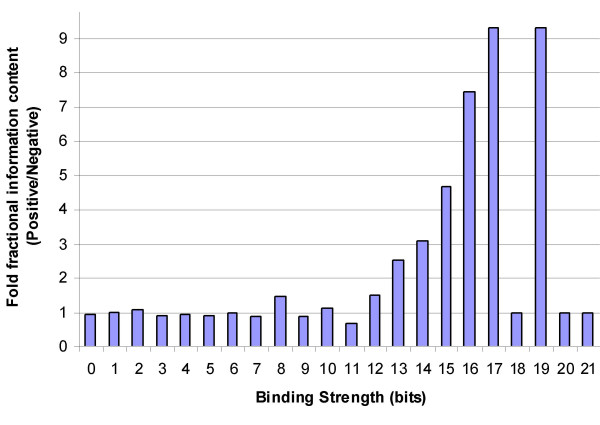
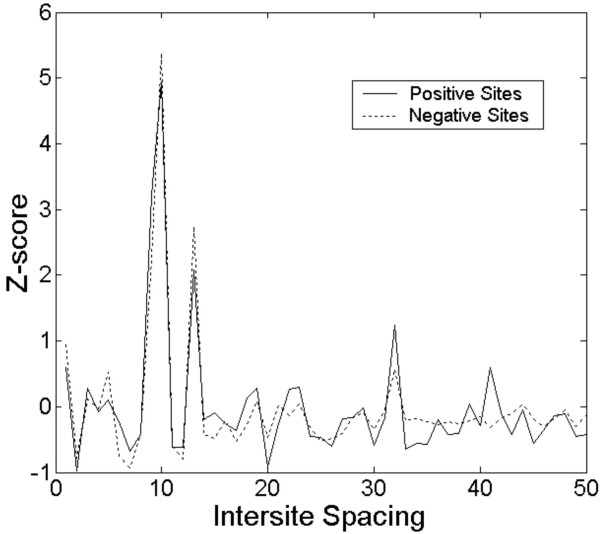
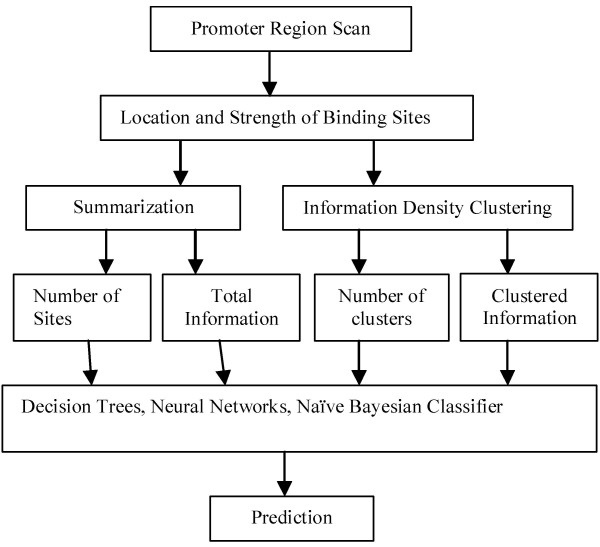
Background: The identification of promoter regions that are regulated by a given transcription factor has traditionally relied upon the identification and distributions of binding sites recognized by the factor. In this study, we have developed a tandem machine learning approach for the identification of regulatory target genes based on these parameters and on the corresponding binding site information contents that measure the affinities of the factor for these cognate elements.
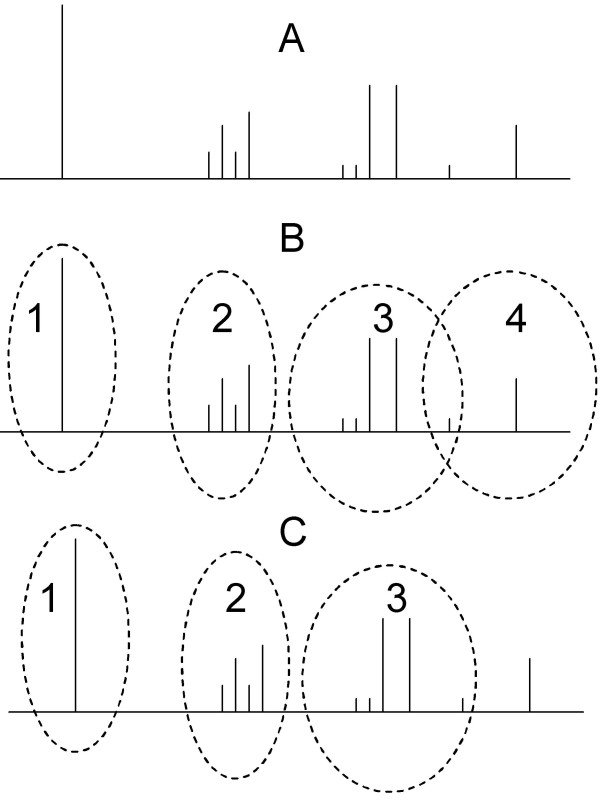
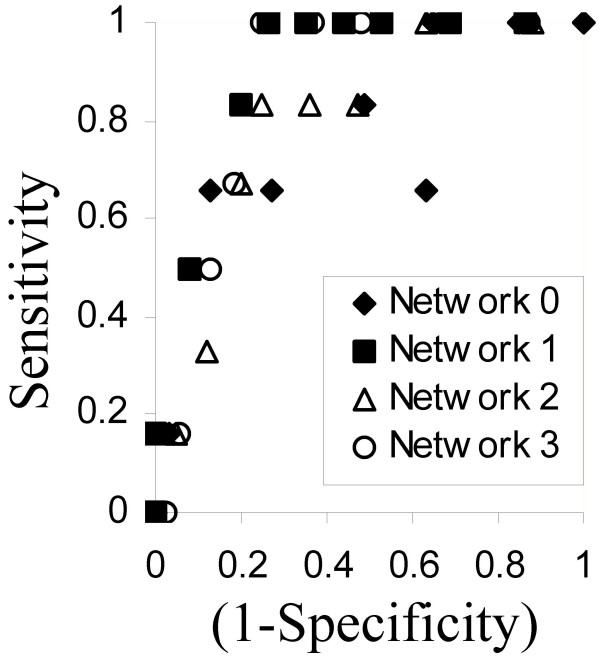
Results: This method has been validated using models of DNA binding sites recognized by the xenobiotic-sensitive nuclear receptor, PXR/RXRalpha, for target genes within the human genome. An information theory-based weight matrix was first derived and refined from known PXR/RXRalpha binding sites. The promoter region of candidate genes was scanned with the weight matrix. A novel information density-based clustering algorithm was then used to identify clusters of information rich sites. Finally, transformed data representing metrics of location, strength and clustering of binding sites were used for classification of promoter regions using an ensemble approach involving neural networks, decision trees and Naïve Bayesian classification. The method was evaluated on a set of 24 known target genes and 288 genes known not to be regulated by PXR/RXRalpha. We report an average accuracy (proportion of correctly classified promoter regions) of 71%, sensitivity of 73%, and specificity of 70%, based on multiple cross-validation and the leave-one-out strategy. The performance on a test set of 13 genes showed that 10 were correctly classified.
Conclusion: We have developed a machine learning approach for the successful detection of gene targets for transcription factors with high accuracy. The method has been validated for the transcription factor PXR/RXRalpha and has the potential to be extended to other transcription factors.
Figures
References
-
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, Kloos DU, Land S, Lewicki-Potapov B, Michael H, Munch R, Reuter I, Rotert S, Saxel H, Scheer M, Thiele S, Wingender E. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. - DOI - PMC - PubMed
-
- Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 2005;33:D192–6. doi: 10.1093/nar/gki069. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
