

GASH: an improved algorithm for maximizing the number of equivalent residues between two protein structures
- PMID: 16146579
- PMCID: PMC1239909
- DOI: 10.1186/1471-2105-6-221
GASH: an improved algorithm for maximizing the number of equivalent residues between two protein structures
Abstract
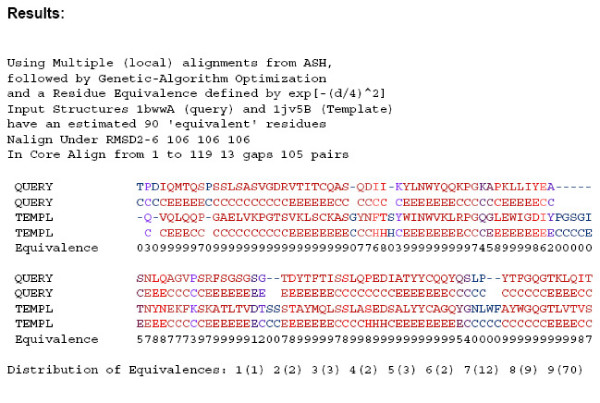
Background: We introduce GASH, a new, publicly accessible program for structural alignment and superposition. Alignments are scored by the Number of Equivalent Residues (NER), a quantitative measure of structural similarity that can be applied to any structural alignment method. Multiple alignments are optimized by conjugate gradient maximization of the NER score within the genetic algorithm framework. Initial alignments are generated by the program Local ASH, and can be supplemented by alignments from any other program.
Results: We compare GASH to DaliLite, CE, and to our earlier program Global ASH on a difficult test set consisting of 3,102 structure pairs, as well as a smaller set derived from the Fischer-Eisenberg set. The extent of alignment crossover, as well as the completeness of the initial set of alignments are examined. The quality of the superpositions is evaluated both by NER and by the number of aligned residues under three different RMSD cutoffs (2,4, and 6A). In addition to the numerical assessment, the alignments for several biologically related structural pairs are discussed in detail.
Conclusion: Regardless of which criteria is used to judge the superposition accuracy, GASH achieves the best overall performance, followed by DaliLite, Global ASH, and CE. In terms of CPU usage, DaliLite CE and GASH perform similarly for query proteins under 500 residues, but for larger proteins DaliLite is faster than GASH or CE. Both an http interface and a simple object application protocol (SOAP) interface to the GASH program are available at http://www.pdbj.org/GASH/.
Figures










References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
