

Constraining ribosomal RNA conformational space
- PMID: 16155182
- PMCID: PMC1214544
- DOI: 10.1093/nar/gki805
Constraining ribosomal RNA conformational space
Abstract
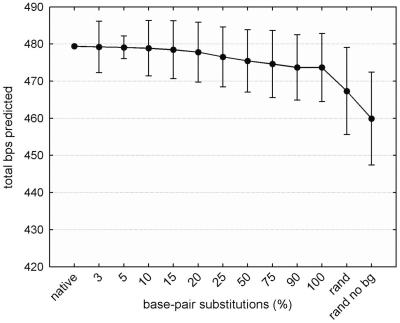
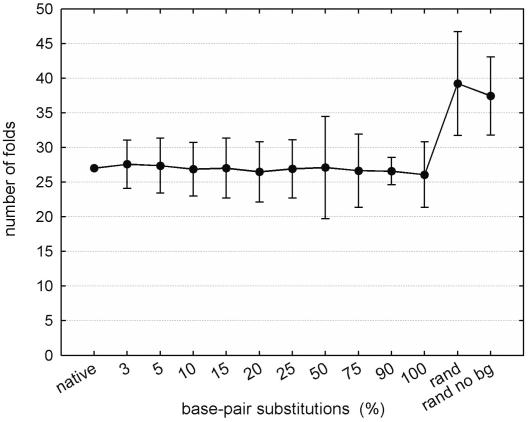
Despite the potential for many possible secondary-structure conformations, the native sequence of ribosomal RNA (rRNA) is able to find the correct and universally conserved core fold. This study reports a computational analysis investigating two mechanisms that appear to constrain rRNA secondary-structure conformational space: ribosomal proteins and rRNA sequence composition. The analysis was carried out by using rRNA-ribosomal protein interaction data for the Escherichia coli 16S rRNA and free energy minimization software for secondary-structure prediction. The results indicate that selection pressures on rRNA sequence composition and ribosomal protein-rRNA interaction play a key role in constraining the rRNA secondary structure to a single stable form.
Figures
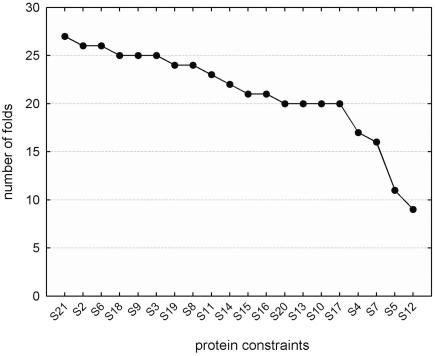
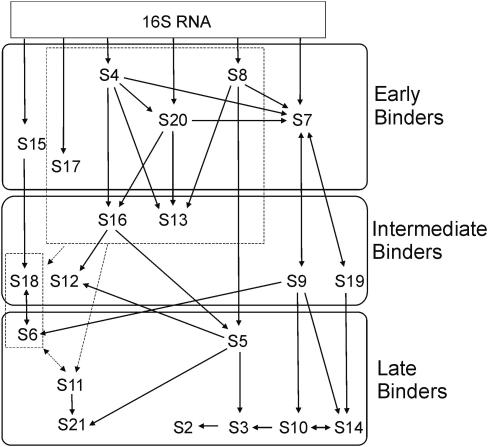
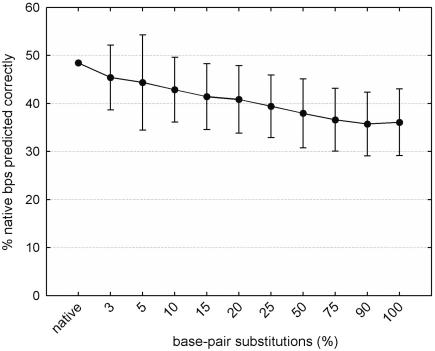
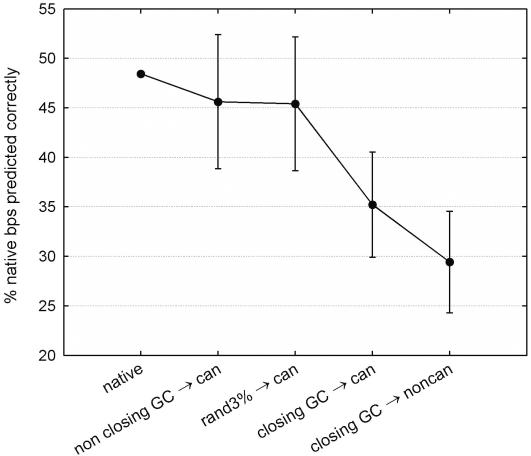
References
-
- Cannone J.J., Subramanian S., Schnare M.N., Collett J.R., D'Souza L.M., Du Y., Feng B., Lin N., Madabusi L.V., Müller K.M., Pande N., Shang Z., Yu N., Gutell R.R. The Comparative RNA Web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. Bioinformatics. 2002;3:2. - PMC - PubMed
-
- Gutell R.R., Weiser B., Woese C.R., Noller H.F. Comparative anatomy of the 16-S-like ribosomal RNA. Prog. Nucleic Acid Res. Mol. Biol. 1985;32:155–216. - PubMed
-
- Brodersen D.E., Clemons W.E., Jr, Carter A.P., Wimberly B.T., Ramakrishnan V. Crystal structure of the 30S ribosomal subunit from Thermus thermophilus: structure of the proteins and their interactions with 16S RNA. J. Mol. Biol. 2002;316:725–768. - PubMed
-
- Klein D., Moore P., Steitz T. The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J. Mol. Biol. 2004;340:141–177. - PubMed
-
- Wimberly B.T., Brodersen D.E., Clemons W.M., Jr, Morgan-Warren R.J., Carter A.P., Vonrhein C., Hartsch T., Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. - PubMed
