

Involvement of PKR and RNase L in translational control and induction of apoptosis after Hepatitis C polyprotein expression from a vaccinia virus recombinant
- PMID: 16156900
- PMCID: PMC1242258
- DOI: 10.1186/1743-422X-2-81
Involvement of PKR and RNase L in translational control and induction of apoptosis after Hepatitis C polyprotein expression from a vaccinia virus recombinant
Abstract
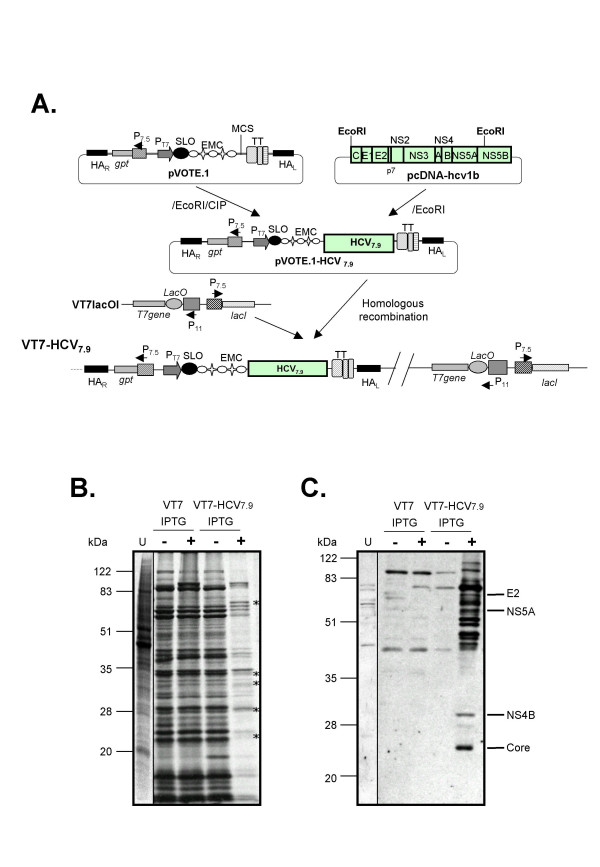
Background: Hepatitis C virus (HCV) infection is of growing concern in public health with around 350 million chronically infected individuals worldwide. Although the IFN-alpha/rivabirin is the only approved therapy with 10-30% clinical efficacy, the protective molecular mechanism involved during the treatment is still unknown. To analyze the effect of HCV polyprotein expression on the antiviral response of the host, we developed a novel vaccinia virus (VV)-based delivery system (VT7-HCV7.9) where structural and nonstructural (except part of NS5B) proteins of HCV ORF from genotype 1b are efficiently expressed and produced, and timely regulated in mammalian cell lines.
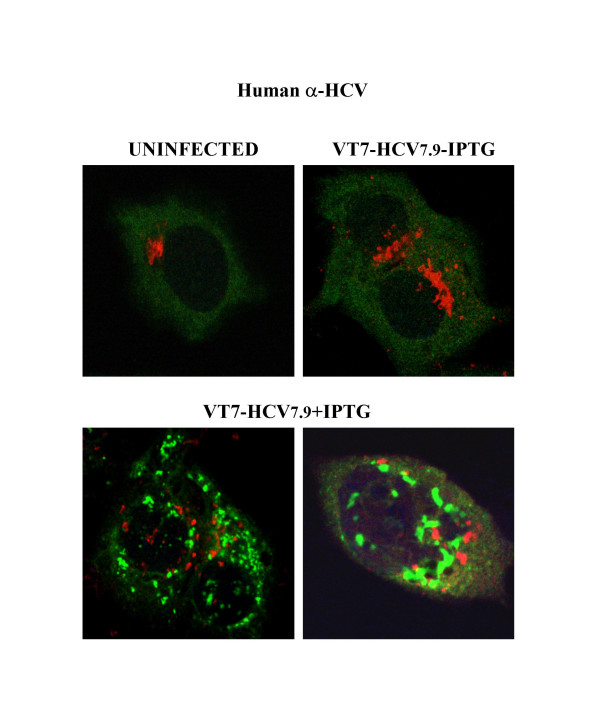
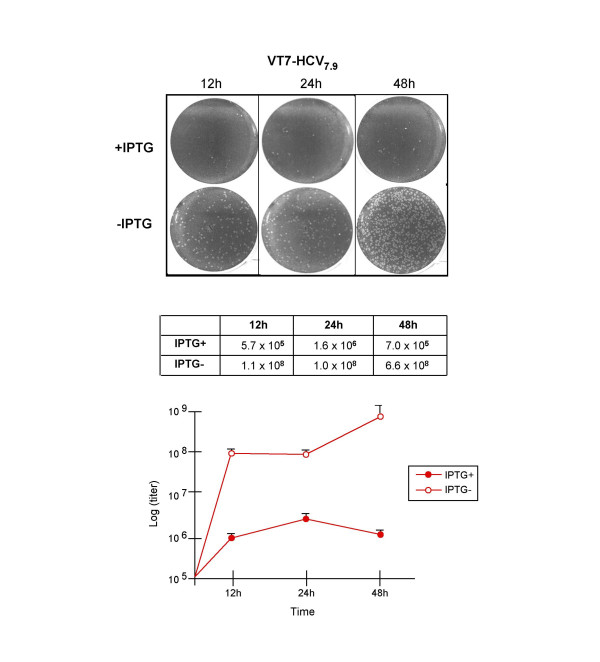
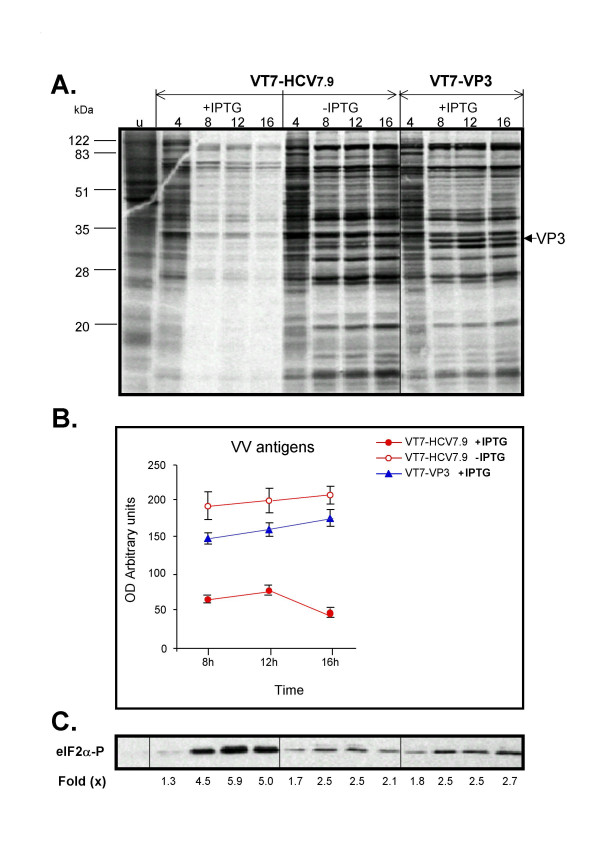
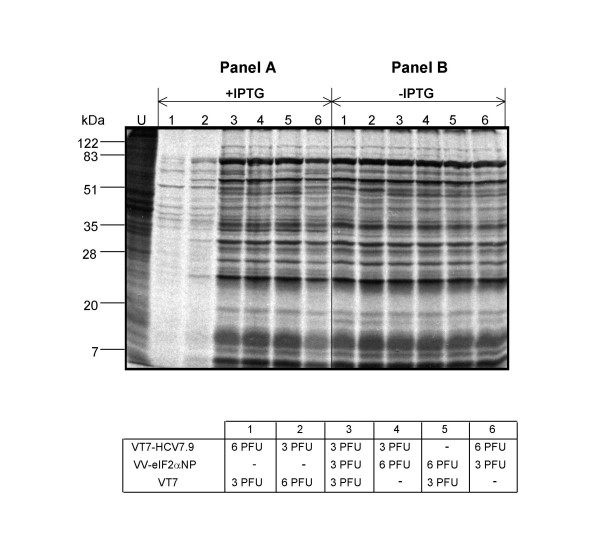
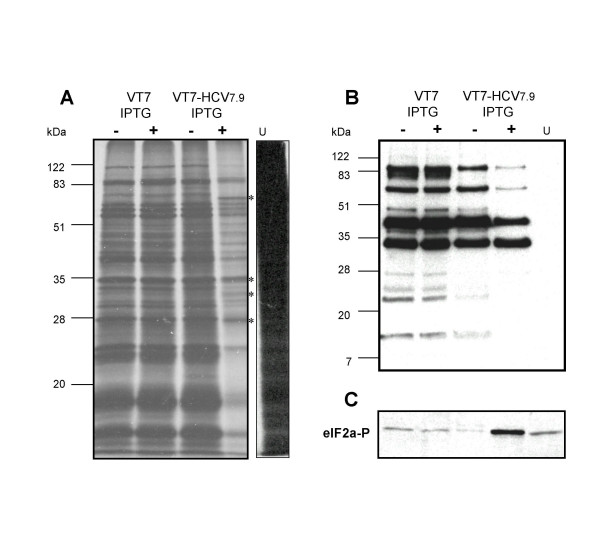
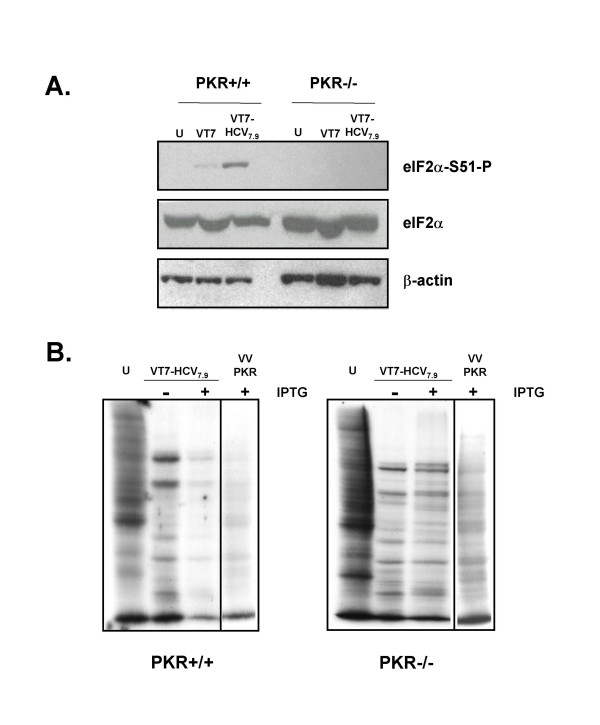
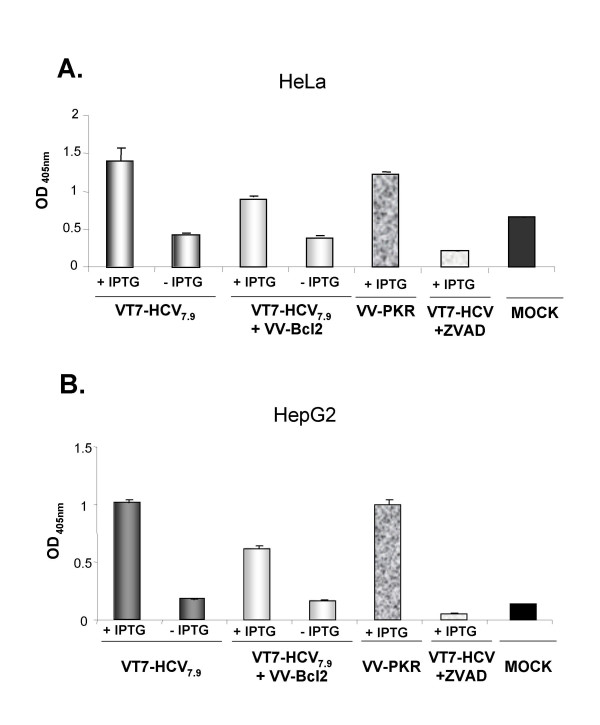
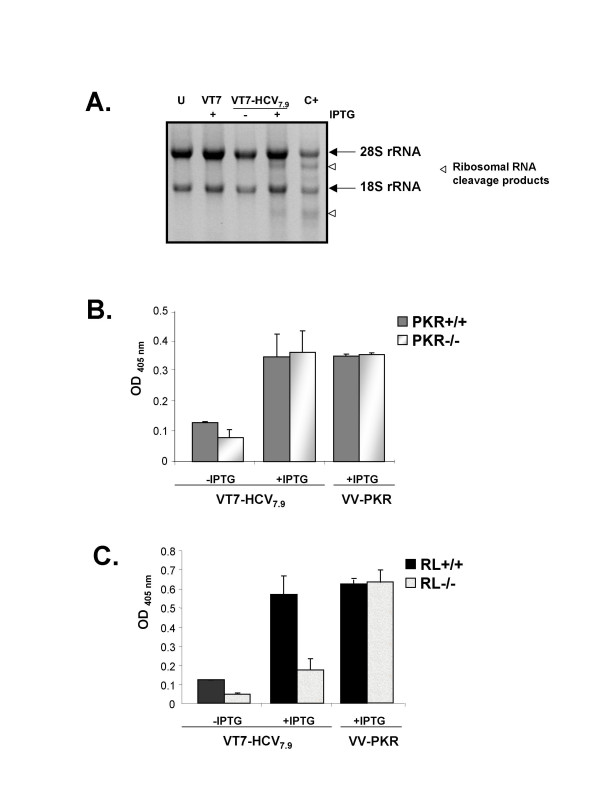
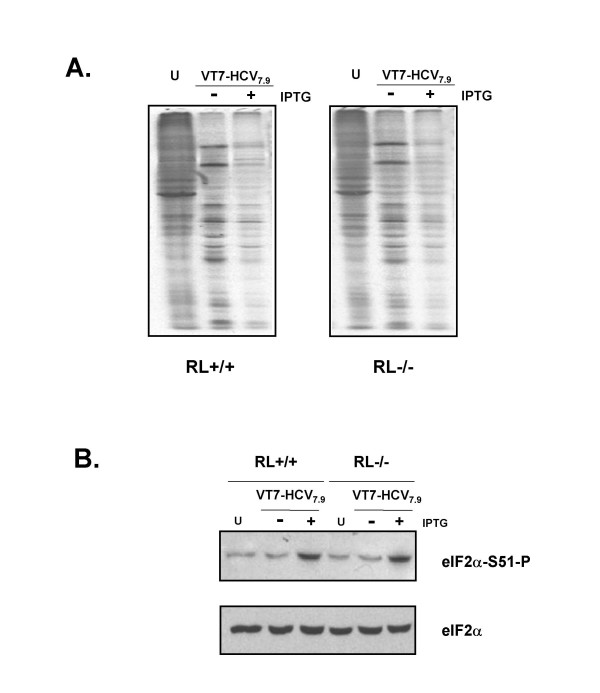
Results: Regulated transcript production and viral polypeptide processing was demonstrated in various cell lines infected with the recombinant VT7-HCV7.9, indicating that the cellular and viral proteolytic machineries are functional within these cells. The inducible expression of the HCV polyprotein by VV inhibits the synthesis of both host and viral proteins over the time and also induces apoptosis in HeLa and HepG2-infected cells. These effects occur accompanying with the phosphorylation of the translation initiation factor eIF-2alpha. In cells co-infected with VT7-HCV7.9 and a recombinant VV expressing the dominant negative eIF-2alpha-S51A mutant in the presence of the inductor isopropyl-thiogalactoside (IPTG), protein synthesis is rescued. The IFN-inducible protein kinase PKR is responsible for the translational block, as demonstrated with PKR-/- and PKR +/+ cell lines. However, apoptosis induced by VT7-HCV7.9 is mediated by the RNase L pathway, in a PKR-independent manner.
Conclusion: These findings demonstrate the antiviral relevance of the proteins induced by interferon, PKR and RNase L during expression from a VV recombinant of the HCV polyprotein in human cell lines. HCV polyprotein expression caused a severe cytopathological effect in human cells as a result of inhibition of protein synthesis and apoptosis induction, triggered by the activation of the IFN-induced enzymes PKR and RNase L systems. Thus, the virus-cell system described here highlights the relevance of the IFN system as a protective mechanism against HCV infection.
Figures
References
-
- Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. - PubMed
-
- Kuo G, Choo QL, Alter HJ, Gitnick GL, Redeker AG, Purcell RH, Miyamura T, Dienstag JL, Alter MJ, Stevens CE, et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244:362–364. - PubMed
-
- Robertson B, Myers G, Howard C, Brettin T, Bukh J, Gaschen B, Gojobori T, Maertens G, Mizokami M, Nainan O, Netesov S, Nishioka K, Shin i T, Simmonds P, Smith D, Stuyver L, Weiner A. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. International Committee on Virus Taxonomy. Arch Virol. 1998;143:2493–2503. doi: 10.1007/s007050050479. - DOI - PubMed
-
- Reed KE, Rice CM. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr Top Microbiol Immunol. 2000;242:55–84. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
