

Towards a genome-based taxonomy for prokaryotes
- PMID: 16159757
- PMCID: PMC1236649
- DOI: 10.1128/JB.187.18.6258-6264.2005
Towards a genome-based taxonomy for prokaryotes
Abstract
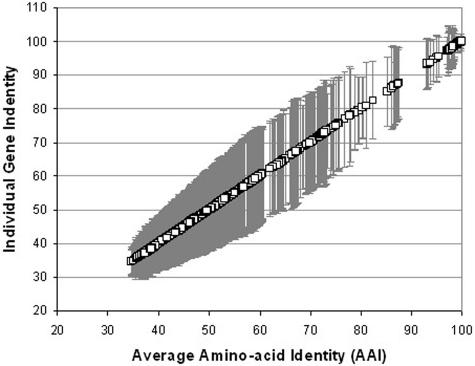
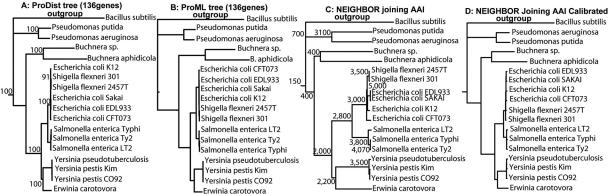
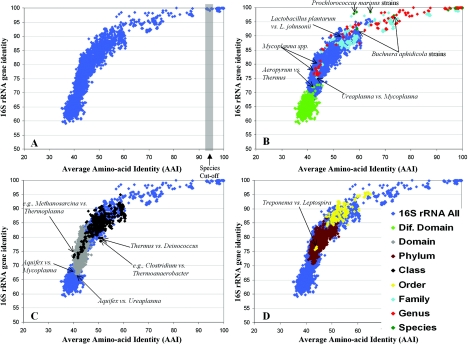
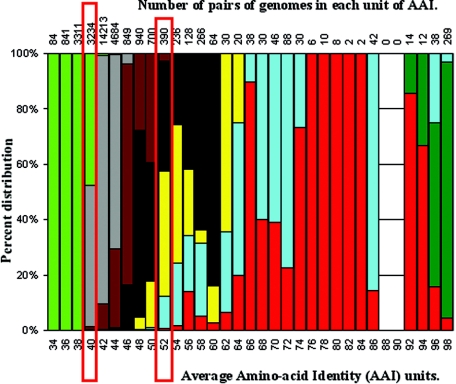
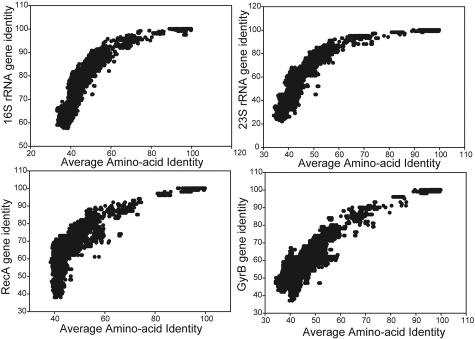
The ranks higher than the species in the prokaryotic taxonomy are primarily designated based on phylogenetic analysis of the 16S rRNA gene sequences, but no definite standards exist for the absolute relatedness (measured by 16S rRNA or other means) between the ranks. Accordingly, it remains unknown how comparable the ranks are between different organisms. To gain insights into this question, we studied the relationship between shared gene content and genetic relatedness for 175 fully sequenced strains, using as a robust measure of relatedness the average amino acid identity (AAI) of the shared genes. Our results reveal that adjacent ranks (e.g., phylum versus class) frequently show extensive overlap in terms of genetic and gene content relatedness of the grouped organisms, and hence, the current system is of limited predictive power in this respect. The overlap between nonadjacent ranks (e.g., phylum versus family) is generally limited and attributable to clear inconsistencies of the taxonomy. In addition to providing means for standardizing taxonomy, our AAI-based approach provides a means to evaluate the robustness of alternative genetic markers for phylogenetic purposes. For instance, the 23S rRNA gene was found to be as good a marker as the 16S rRNA gene, while several of the widely distributed protein-coding genes, such as the RNA polymerase and gyrase subunits, show a strong phylogenetic signal, albeit less strong than the rRNA genes (0.78 > R2 > 0.69 for the protein-coding genes versus R2 = 0.84 for the rRNA genes). The AAI approach outlined here could contribute significantly to a genome-based taxonomy for all microbial organisms.
Figures
Comment in
-
Updating prokaryotic taxonomy.J Bacteriol. 2005 Sep;187(18):6255-7. doi: 10.1128/JB.187.18.6255-6257.2005. J Bacteriol. 2005. PMID: 16159756 Free PMC article. No abstract available.
References
-
- Brenner, D., J. Staley, and N. Krieg. 2000. Classification of prokaryotic organisms and the concept of bacterial speciation, p. 27-31. In D. R. Boone, R. W. Castenholz, and G. M. Garrity (ed.), Bergey's manual of systematic bacteriology, 2nd ed., vol. 1. Springer-Verlag, New York, N.Y.
-
- Brown, J. R., C. J. Douady, M. J. Italia, W. E. Marshall, and M. J. Stanhope. 2001. Universal trees based on large combined protein sequence data sets. Nat. Genet. 28:281-285. - PubMed
-
- Canale-Parola, E. 1984. Order I: Spirochaetales Buchanan 1917, 163AL, p. 38-39. In N. R. Krieg, N. and J. G. Holt (ed.), Bergey's manual of systematic bacteriology, vol. 1. William and Wilkins, Baltimore, Md.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
