

Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis
- PMID: 16162919
- PMCID: PMC6725659
- DOI: 10.1523/JNEUROSCI.2164-05.2005
Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis
Abstract
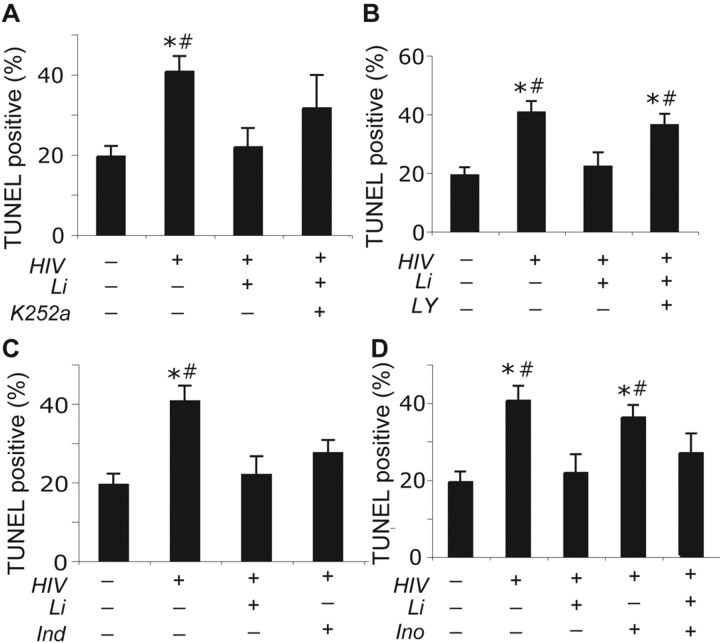
Lithium (Li) has garnered considerable interest as a neuroprotective drug for a broad range of nervous system disorders. Its neuroprotective activities occur as a consequence of glycogen synthase kinase-3beta (GSK-3beta) inhibition leading to downstream blockade of beta-catenin and Tau phosphorylation. In the present study, we investigated Li-mediated neuroprotective mechanisms in laboratory and murine human immunodeficiency virus-1 (HIV-1) encephalitis (HIVE) models. In laboratory tests, Li protected neurons from neurotoxic secretions of HIV-1-infected monocyte-derived macrophages (MDMs). This neuroprotection was mediated, in part, through the phosphatidyl inositol 3-kinase/Akt and GSK-3beta pathways. To examine the effects of Li treatment in vivo, MDMs were injected into the basal ganglia of severe combined immunodeficient mice and then Li was administered (60 mg/kg/d). Seven days after MDM injection, mice were killed and CNS tissue was collected and subjected to immunocytochemical and Western blot assays for leukocyte and neural antigens, GSK-3beta, and key kinase substrates such as beta-catenin and Tau. Numbers of HIV-1 p24 antigen-positive MDMs were unaltered by Li treatment of HIVE mice. Similarly, the greatly increased extent of astrocyte and microglia activation in HIVE mice (10-fold and 16-fold, respectively, compared with unmanipulated controls) was also unaltered by Li. In contrast, Li restored HIVE-associated loss of microtubule-associated protein-2-positive neurites and synaptic density while reducing levels or activity of phospho-Tau Ser202, phospho-beta-catenin, and GSK-3beta. Electrophysiological recordings showed diminished long-term potentiation in hippocampal slices of HIVE mice that were restored by Li. Based on these data, the use of Li as an adjuvant for HIV-1-associated dementia is now being pursued.
Figures








References
-
- Anderson ER, Boyle J, Zink WE, Persidsky Y, Gendelman HE, Xiong H (2003) Hippocampal synaptic dysfunction in a murine model of human immunodeficiency virus type 1 encephalitis. Neuroscience 118: 359–369. - PubMed
-
- Berry GT, Buccafusca R, Greer JJ, Eccleston E (2004) Phosphoinositide deficiency due to inositol depletion is not a mechanism of lithium action in brain. Mol Genet Metab 82: 87–92. - PubMed
-
- Bhat RV, Budd Haeberlein SL, Avila J (2004) Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem 89: 1313–1317. - PubMed
-
- Bodner A, Toth PT, Miller RJ (2004) Activation of c-Jun N-terminal kinase mediates gp120IIIB- and nucleoside analogue-induced sensory neuron toxicity. Exp Neurol 188: 246–253. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical