

Evidence of a large-scale functional organization of mammalian chromosomes
- PMID: 16163395
- PMCID: PMC1201368
- DOI: 10.1371/journal.pgen.0010033
Evidence of a large-scale functional organization of mammalian chromosomes
Abstract
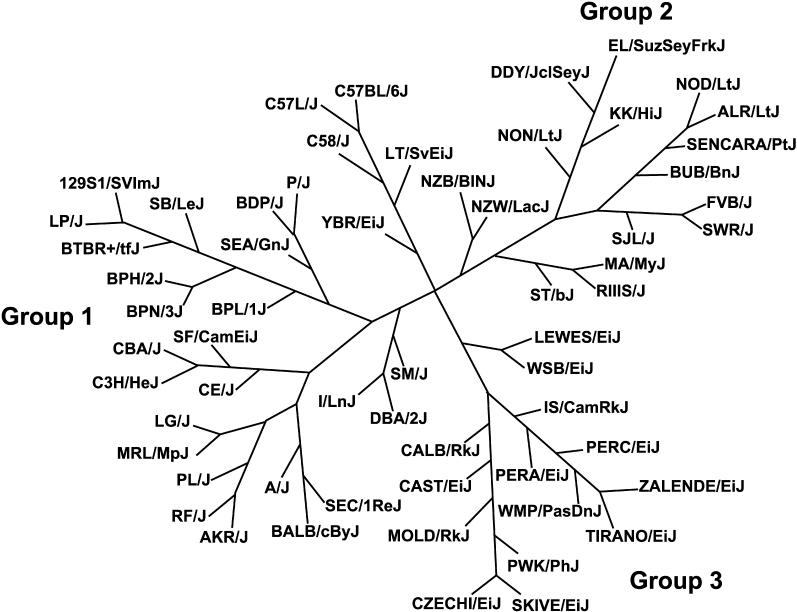
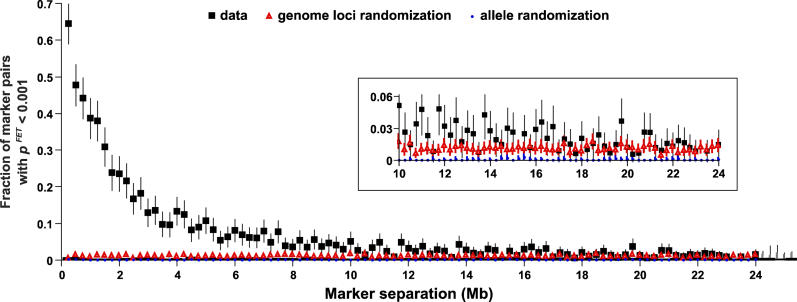
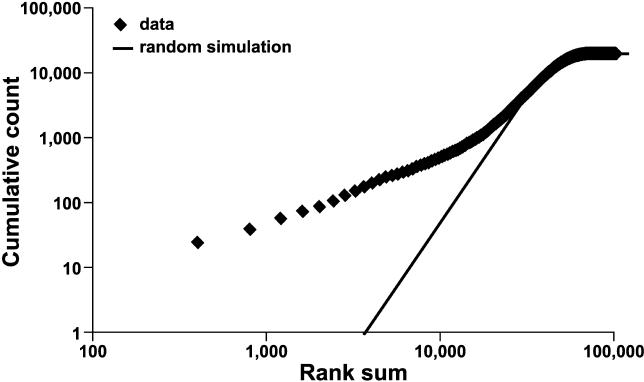
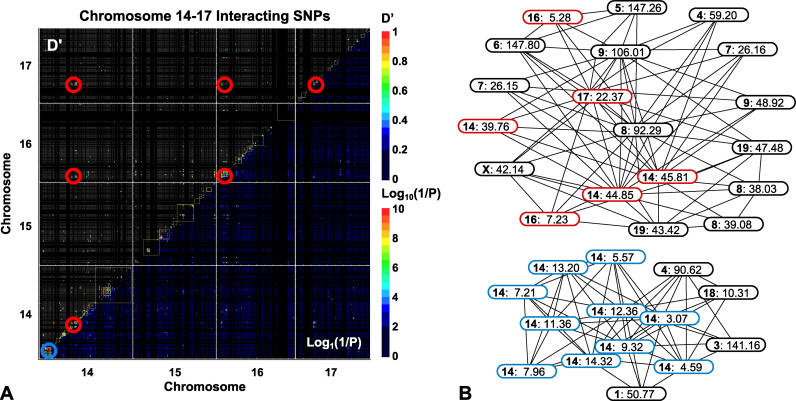
Evidence from inbred strains of mice indicates that a quarter or more of the mammalian genome consists of chromosome regions containing clusters of functionally related genes. The intense selection pressures during inbreeding favor the coinheritance of optimal sets of alleles among these genetically linked, functionally related genes, resulting in extensive domains of linkage disequilibrium (LD) among a set of 60 genetically diverse inbred strains. Recombination that disrupts the preferred combinations of alleles reduces the ability of offspring to survive further inbreeding. LD is also seen between markers on separate chromosomes, forming networks with scale-free architecture. Combining LD data with pathway and genome annotation databases, we have been able to identify the biological functions underlying several domains and networks. Given the strong conservation of gene order among mammals, the domains and networks we find in mice probably characterize all mammals, including humans.
Conflict of interest statement
Competing interests. The authors have declared that no competing interests exist.
Figures



References
-
- Hurst LD, Pal C, Lercher MJ. The evolutionary dynamics of eukaryotic gene order. Nat Rev Genet. 2004;5:299–310. - PubMed
-
- Fisher RA. The genetical theory of natural selection. Oxford, United Kingdom: Clarendon Press; 1930. 318. p.
-
- Nei M. Genome evolution: Let's stick together. Heredity. 2003;90:411–412. - PubMed
-
- Dobzhansky T. Genetics of the evolutionary process. New York: Columbia University Press; 1970. 505. p.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
