

Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species
- PMID: 16166634
- PMCID: PMC1265746
- DOI: 10.1128/MCB.25.19.8520-8530.2005
Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species
Abstract
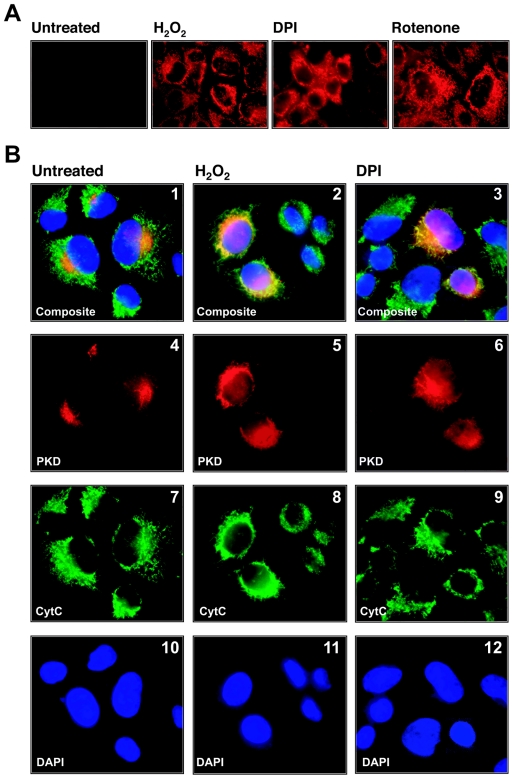
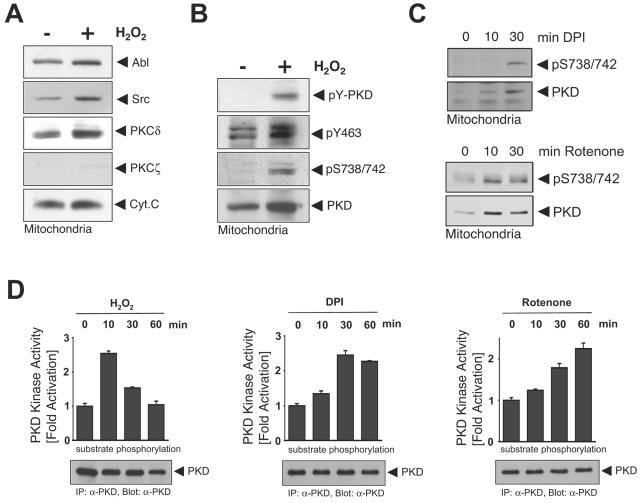
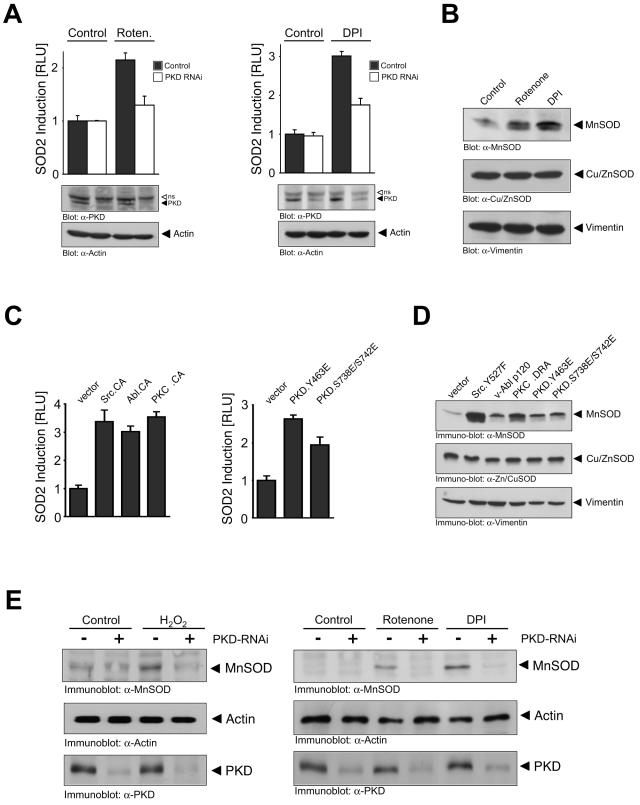
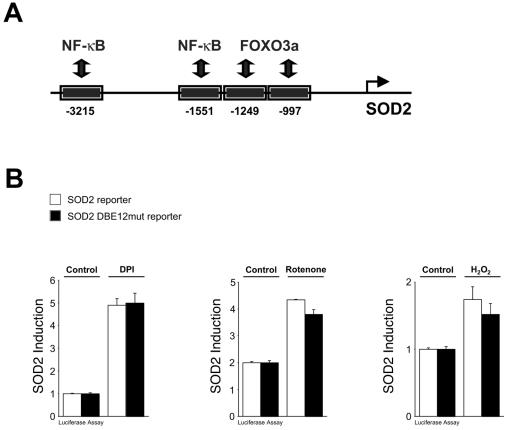
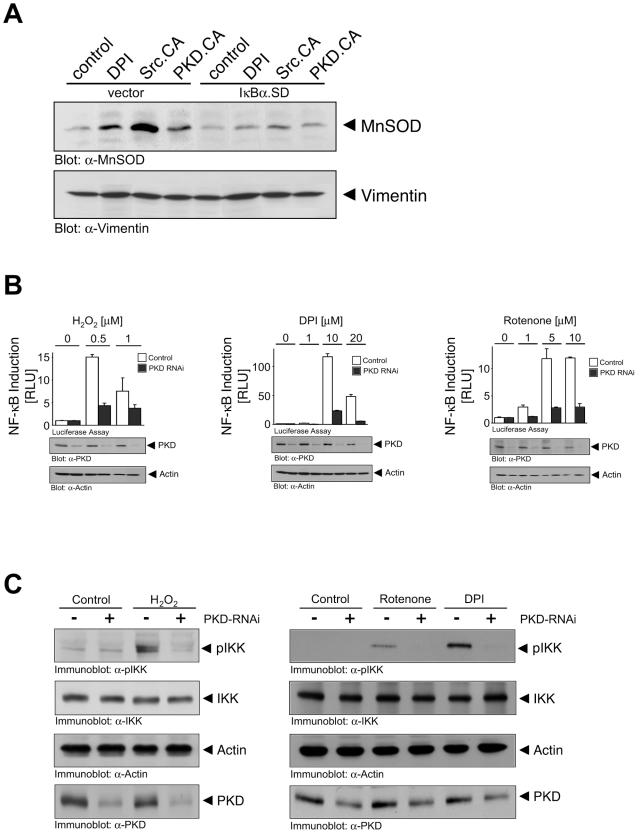
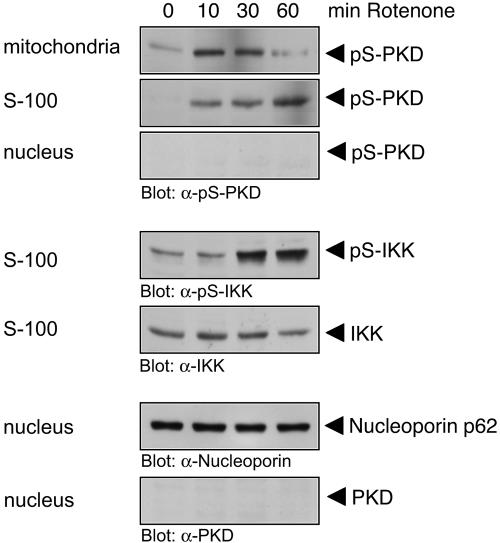
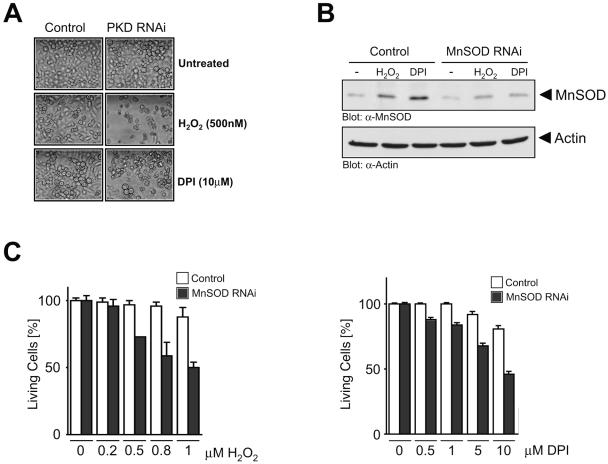
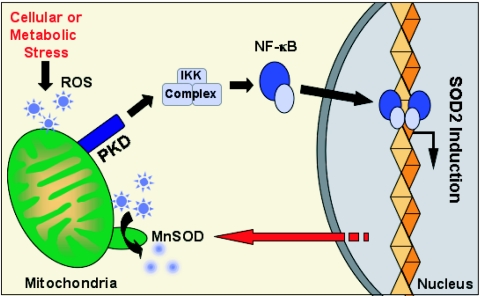
Efficient elimination of mitochondrial reactive oxygen species (mROS) correlates with increased cellular survival and organism life span. Detoxification of mitochondrial ROS is regulated by induction of the nuclear SOD2 gene, which encodes the manganese-dependent superoxide dismutase (MnSOD). However, the mechanisms by which mitochondrial oxidative stress activates cellular signaling pathways leading to induction of nuclear genes are not known. Here we demonstrate that release of mROS activates a signal relay pathway in which the serine/threonine protein kinase D (PKD) activates the NF-kappaB transcription factor, leading to induction of SOD2. Conversely, the FOXO3a transcription factor is dispensable for mROS-induced SOD2 induction. PKD-mediated MnSOD expression promotes increased survival of cells upon release of mROS, suggesting that mitochondrion-to-nucleus signaling is necessary for efficient detoxification mechanisms and cellular viability.
Figures
References
-
- Barnham, K. J., C. L. Masters, and A. I. Bush. 2004. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 3:205-214. - PubMed
-
- Brummelkamp, T. R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science 296:550-553. - PubMed
-
- Brunet, A., A. Bonni, M. J. Zigmond, M. Z. Lin, P. Juo, L. S. Hu, M. J. Anderson, K. C. Arden, J. Blenis, and M. E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857-868. - PubMed
-
- Brunet, A., L. B. Sweeney, J. F. Sturgill, K. F. Chua, P. L. Greer, Y. Lin, H. Tran, S. E. Ross, R. Mostoslavsky, H. Y. Cohen, L. S. Hu, H. L. Cheng, M. P. Jedrychowski, S. P. Gygi, D. A. Sinclair, F. W. Alt, and M. E. Greenberg. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011-2015. - PubMed
-
- Finkel, T., and N. J. Holbrook. 2000. Oxidants, oxidative stress and the biology of ageing. Nature 408:239-247. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials