

Direct observation of thymine dimer repair in DNA by photolyase
- PMID: 16169906
- PMCID: PMC1283438
- DOI: 10.1073/pnas.0506586102
Direct observation of thymine dimer repair in DNA by photolyase
Abstract
Photolyase uses light energy to split UV-induced cyclobutane dimers in damaged DNA, but its molecular mechanism has never been directly revealed. Here, we report the direct mapping of catalytic processes through femtosecond synchronization of the enzymatic dynamics with the repair function. We observed direct electron transfer from the excited flavin cofactor to the dimer in 170 ps and back electron transfer from the repaired thymines in 560 ps. Both reactions are strongly modulated by active-site solvation to achieve maximum repair efficiency. These results show that the photocycle of DNA repair by photolyase is through a radical mechanism and completed on subnanosecond time scale at the dynamic active site, with no net change in the redox state of the flavin cofactor.
Figures
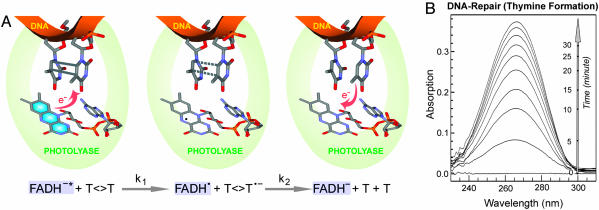
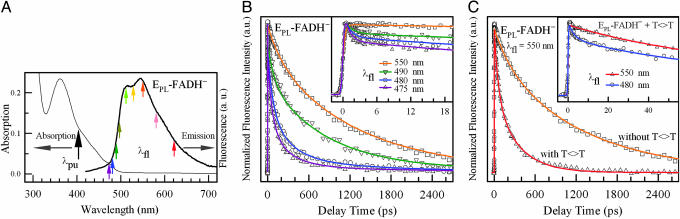
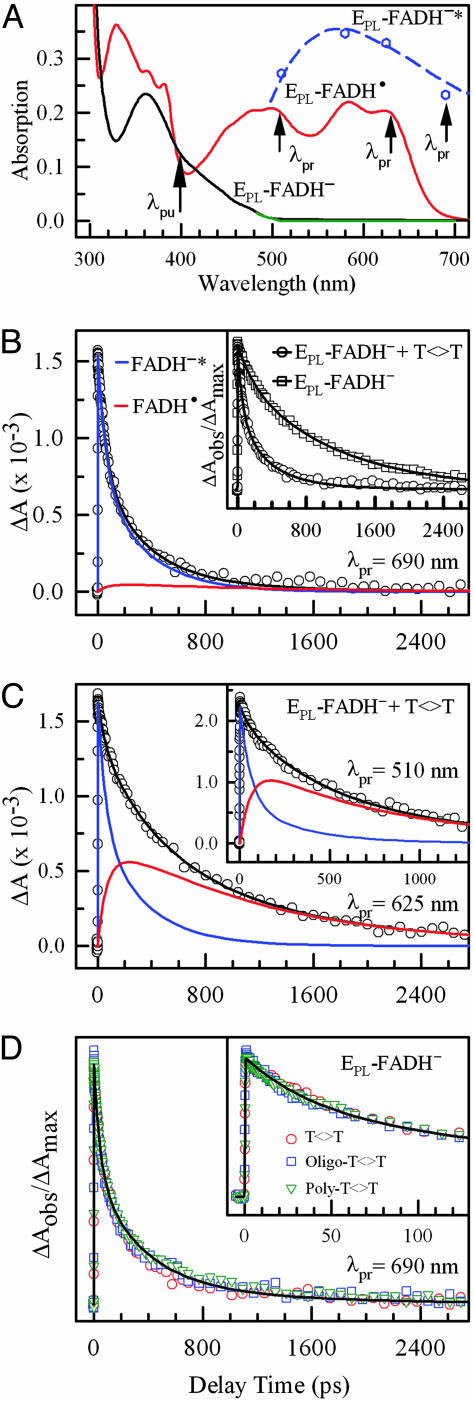
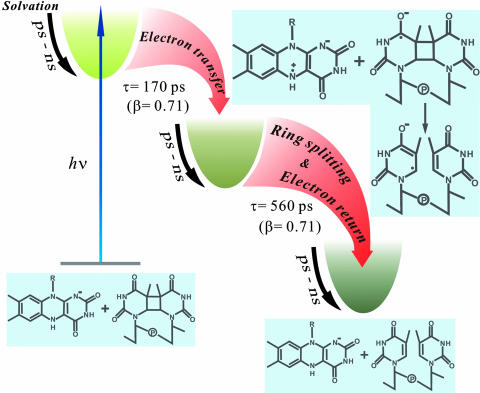
References
-
- Brash, D. E. (1997) Trends Genet. 13, 410–414. - PubMed
-
- Sancar, A. (2003) Chem. Rev. 103, 2203–2237. - PubMed
-
- Park, H. W., Kim, S. T., Sancar, A. & Deisenhofer, J. (1995) Science 268, 1866–1872. - PubMed
-
- Sancar, G. B., Jorns, M. S., Payne, G., Fluke, D. J., Rupert, C. S. & Sancar, A. (1987) J. Biol. Chem. 262, 492–498. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
