

Long-term hydroxyurea therapy for infants with sickle cell anemia: the HUSOFT extension study
- PMID: 16172253
- PMCID: PMC1895275
- DOI: 10.1182/Blood-2004-12-4973
Long-term hydroxyurea therapy for infants with sickle cell anemia: the HUSOFT extension study
Abstract
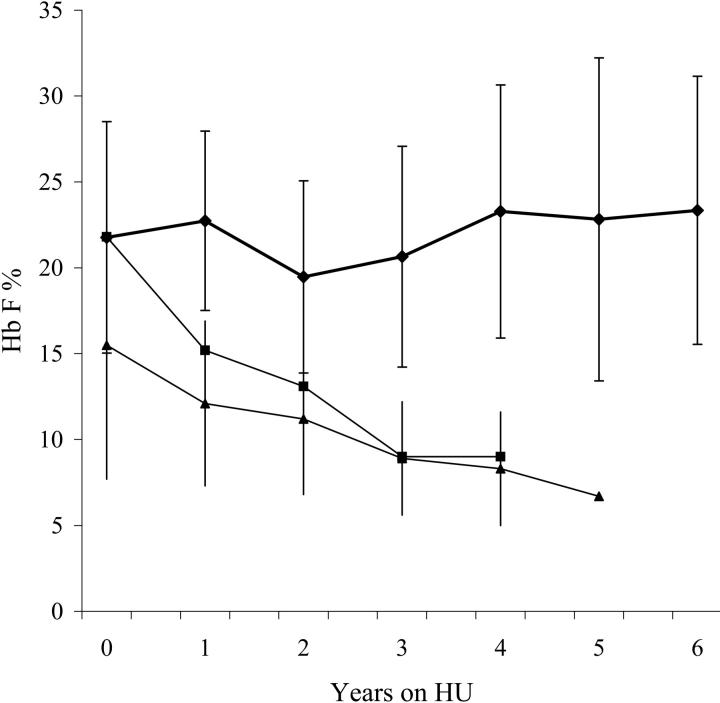
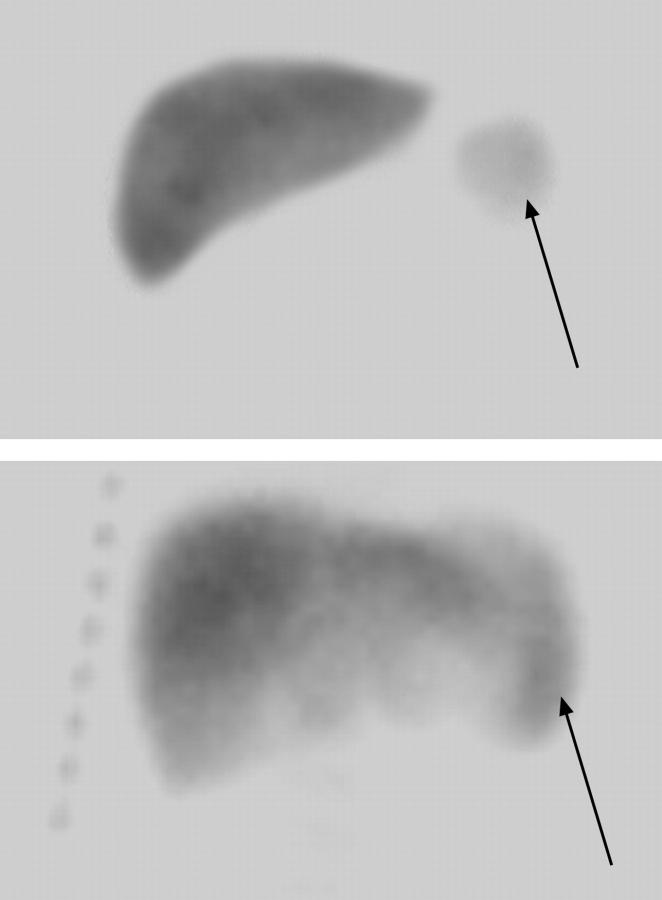
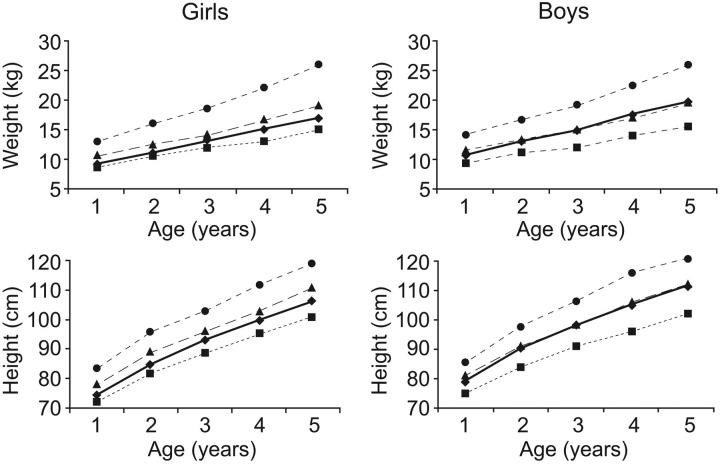
The long-term efficacy and toxicity of hydroxyurea for infants are undefined, and its role in preventing organ dysfunction is unknown. Short-term feasibility of hydroxyurea administration, toxicities, hematologic effects, and effect on spleen function in infants with sickle cell anemia (SCA) were reported (Hydroxyurea Safety and Organ Toxicity [HUSOFT] trial). These infants completing 2 years of hydroxyurea therapy (20 mg/kg/d) were offered study extension with dose escalation to 30 mg/kg/d. Patients were monitored with laboratory tests and biannual imaging studies. Hematologic indices were compared with predicted age-specific values and event rates compared with historic rates. All 21 subjects completing the original trial enrolled in the extension study: median age, 3.4 years old (range, 2.6 to 4.4 years); 12 females; 20 with Hb SS, 1 with Hb S/beta0-thalassemia. Seventeen patients completed 4 years of hydroxyurea, and 11 completed 6 years. After 4 years, hydroxyurea was associated with increased hemoglobin concentration, percentage of fetal hemoglobin (Hb F), and mean corpuscular volume (MCV) and decreased reticulocytes, white blood cells (WBCs), and platelets (P < .01). Patients experienced 7.5 acute chest syndrome (ACS) events per 100 person-years, compared with 24.5 events per 100 person-years among historic controls (P = .001). Treated patients had better spleen function than expected and improved growth rates. Infants with SCA tolerate prolonged hydroxyurea therapy with sustained hematologic benefits, fewer ACS events, improved growth, and possibly preserved organ function.
Figures
References
-
- Pearson HA, Spencer RP, Cornelius EA. Functional asplenia in sickle-cell anemia. N Engl J Med. 1969;281: 923-926. - PubMed
-
- Wigfall DR, Ware RE, Burchinal MR, Kinney TR, Foreman JW. Prevalence and clinical correlates of glomerulopathy in children with sickle cell disease. J Pediatr. 2000;136: 749-753. - PubMed
-
- Powars D, Weidman JA, Odom-Maryon T, Niland JC, Johnson C. Sickle cell chronic lung disease: prior morbidity and the risk of pulmonary failure. Medicine (Baltimore). 1988;67: 66-76. - PubMed
-
- Pegelow CH, Macklin EA, Moser FG, et al. Longitudinal changes in brain magnetic resonance imaging findings in children with sickle cell disease. Blood. 2002;99: 3014-3018. - PubMed
-
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330: 1639-1644. - PubMed
