

5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) attenuates the expression of LPS- and Abeta peptide-induced inflammatory mediators in astroglia
- PMID: 16174294
- PMCID: PMC1262754
- DOI: 10.1186/1742-2094-2-21
5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) attenuates the expression of LPS- and Abeta peptide-induced inflammatory mediators in astroglia
Abstract
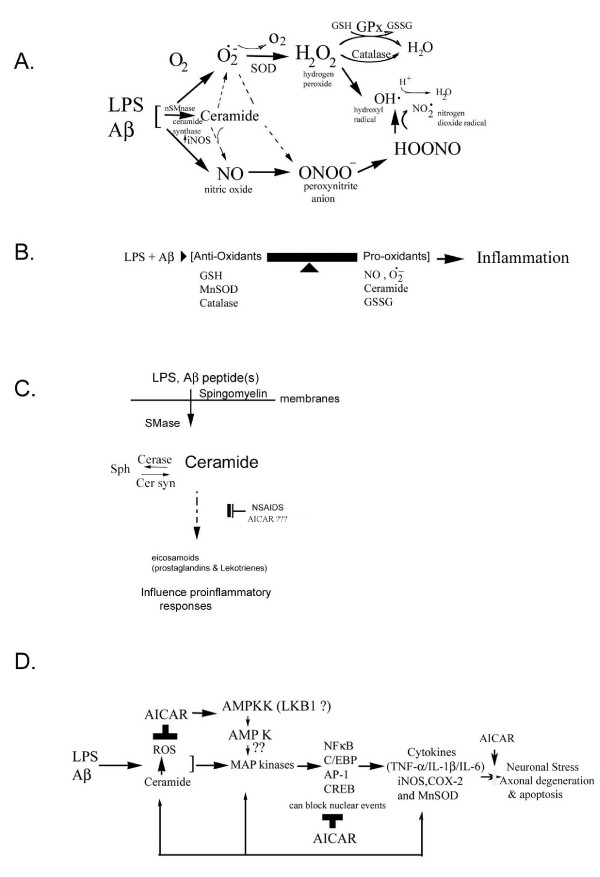
Background: Alzheimer's disease (AD) pathology shows characteristic 'plaques' rich in amyloid beta (Abeta) peptide deposits. Inflammatory process-related proteins such as pro-inflammatory cytokines have been detected in AD brain suggesting that an inflammatory immune reaction also plays a role in the pathogenesis of AD. Glial cells in culture respond to LPS and Abeta stimuli by upregulating the expression of cytokines TNF-alpha, IL-1beta, and IL-6, and also the expression of proinflammatory genes iNOS and COX-2. We have earlier reported that LPS/Abeta stimulation-induced ceramide and ROS generation leads to iNOS expression and nitric oxide production in glial cells. The present study was undertaken to investigate the neuroprotective function of AICAR (a potent activator of AMP-activated protein kinase) in blocking the pro-oxidant/proinflammatory responses induced in primary glial cultures treated with LPS and Abeta peptide.
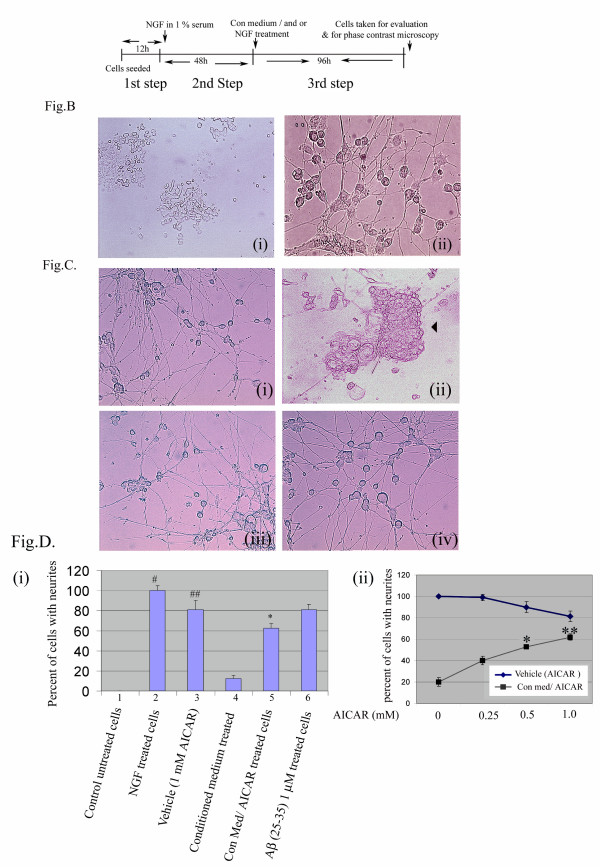
Methods: To test the anti-inflammatory/anti-oxidant functions of AICAR, we tested its inhibitory potential in blocking the expression of pro-inflammatory cytokines and iNOS, expression of COX-2, generation of ROS, and associated signaling following treatment of glial cells with LPS and Abeta peptide. We also investigated the neuroprotective effects of AICAR against the effects of cytokines and inflammatory mediators (released by the glia), in blocking neurite outgrowth inhibition, and in nerve growth factor-(NGF) induced neurite extension by PC-12 cells.
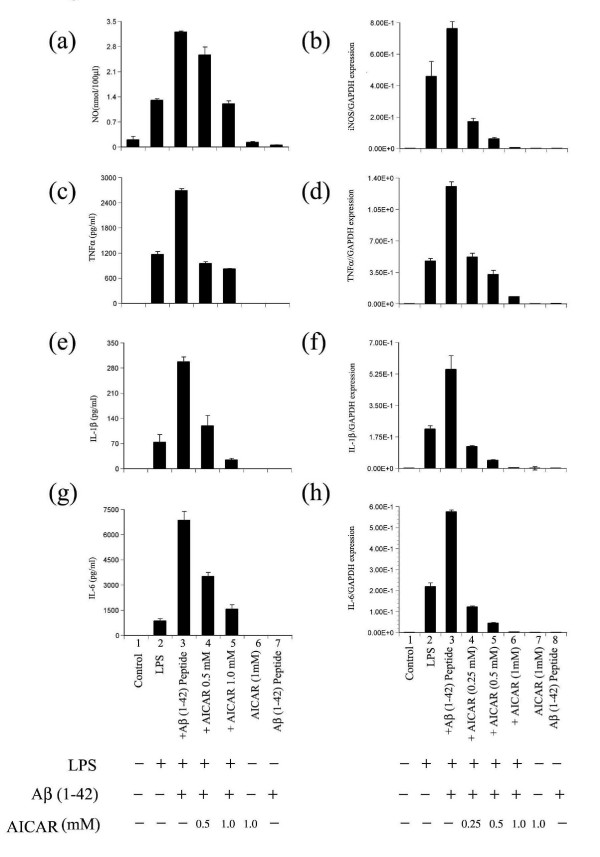
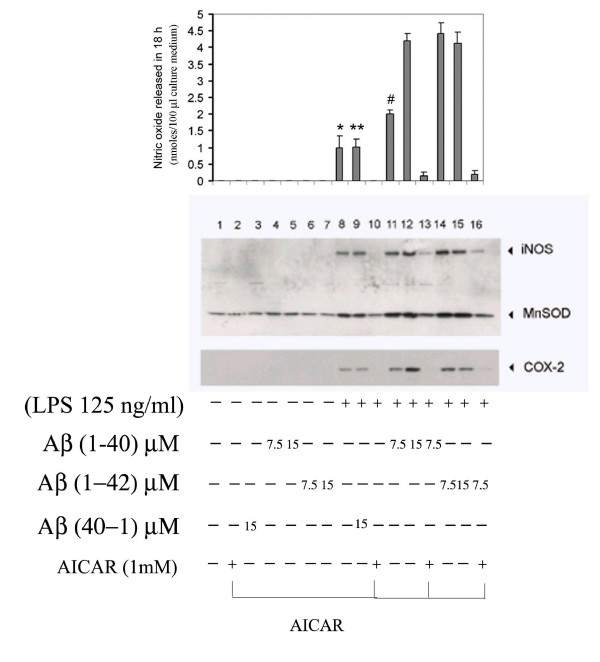
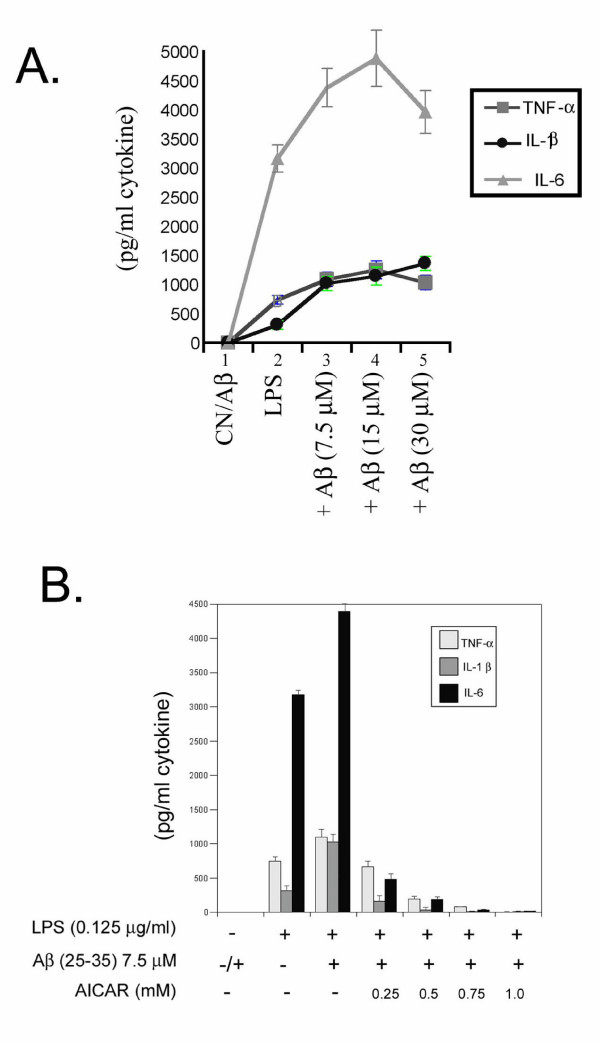
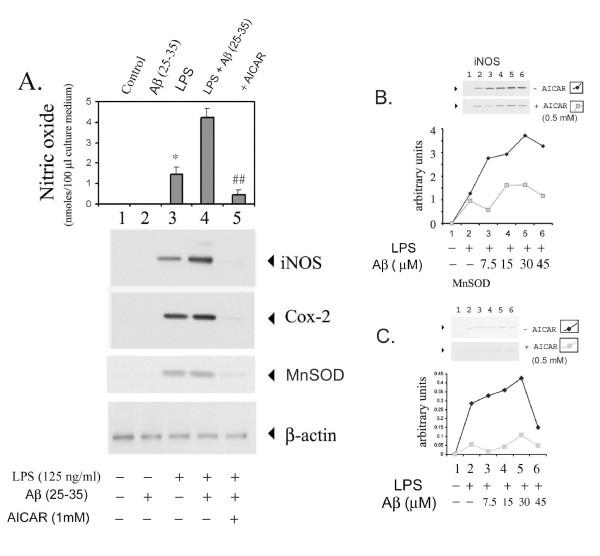
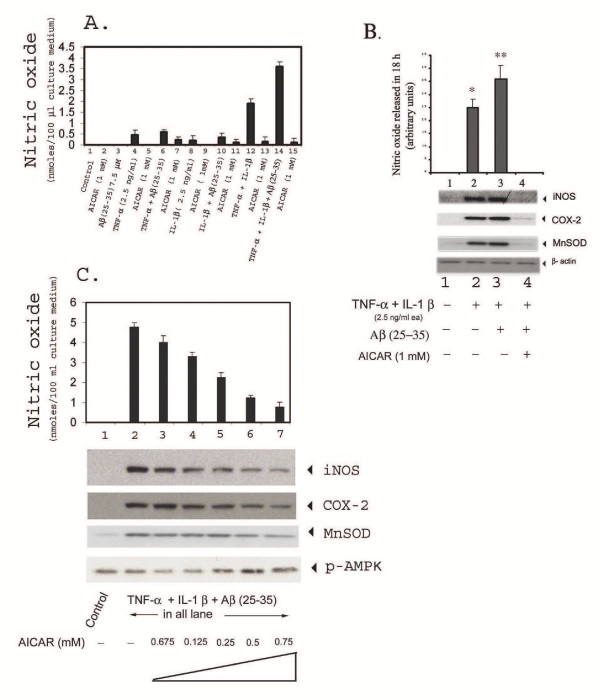
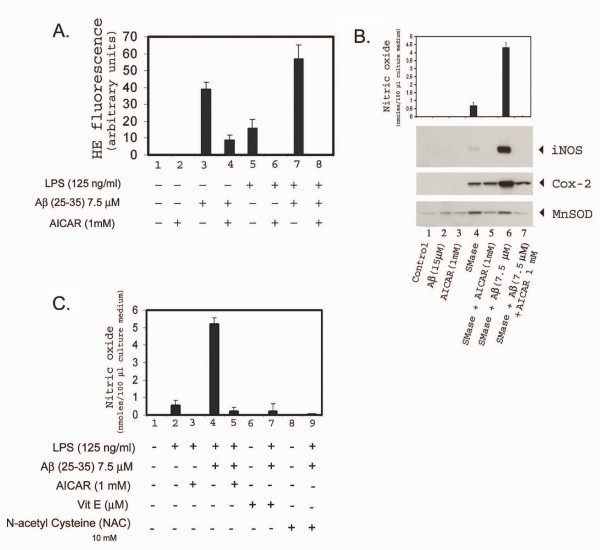
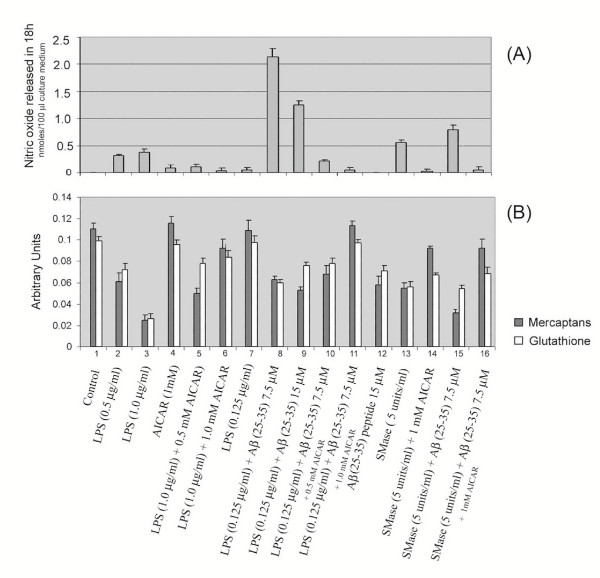
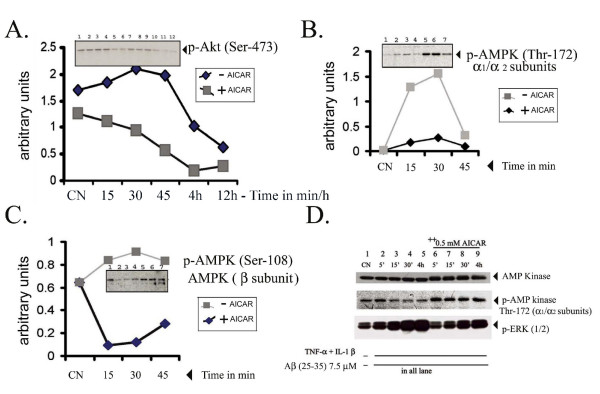
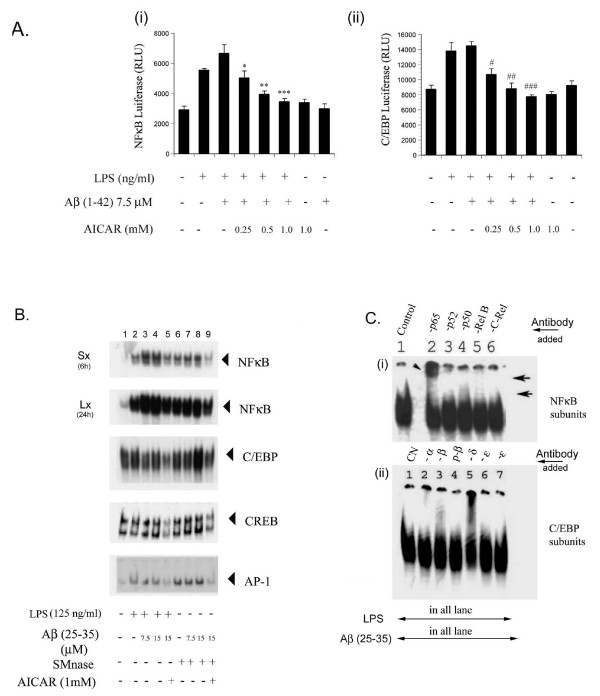
Results: AICAR blocked LPS/Abeta-induced inflammatory processes by blocking the expression of proinflammatory cytokine, iNOS, COX-2 and MnSOD genes, and by inhibition of ROS generation and depletion of glutathione in astroglial cells. AICAR also inhibited down-stream signaling leading to the regulation of transcriptional factors such as NFkappaB and C/EBP which are critical for the expression of iNOS, COX-2, MnSOD and cytokines (TNF-alpha/IL-1beta and IL-6). AICAR promoted NGF-induced neurite growth and reduced neurite outgrowth inhibition in PC-12 cells treated with astroglial conditioned medium.
Conclusion: The observed anti-inflammatory/anti-oxidant and neuroprotective functions of AICAR suggest it as a viable candidate for use in treatment of Alzheimer's disease.
Figures
Similar articles
-
AICAR (5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside) increases the production of toxic molecules and affects the profile of cytokines release in LPS-stimulated rat primary microglial cultures.Neurotoxicology. 2010 Jan;31(1):134-46. doi: 10.1016/j.neuro.2009.10.006. Epub 2009 Oct 22. Neurotoxicology. 2010. PMID: 19853624
-
Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 gene expression by 5-aminoimidazole-4-carboxamide riboside is independent of AMP-activated protein kinase.J Cell Biochem. 2008 Feb 15;103(3):931-40. doi: 10.1002/jcb.21466. J Cell Biochem. 2008. PMID: 17615555
-
Implication of S-adenosylhomocysteine hydrolase in inhibition of TNF-alpha- and IL-1beta-induced expression of inflammatory mediators by AICAR in RPE cells.Invest Ophthalmol Vis Sci. 2008 Mar;49(3):1274-81. doi: 10.1167/iovs.07-1109. Invest Ophthalmol Vis Sci. 2008. PMID: 18326758
-
Piper sarmentosum Roxb. confers neuroprotection on beta-amyloid (Aβ)-induced microglia-mediated neuroinflammation and attenuates tau hyperphosphorylation in SH-SY5Y cells.J Ethnopharmacol. 2018 May 10;217:187-194. doi: 10.1016/j.jep.2018.02.025. Epub 2018 Feb 17. J Ethnopharmacol. 2018. PMID: 29462698
-
Pharmacological Activities and Applications of Spicatoside A.Biomol Ther (Seoul). 2016 Sep 1;24(5):469-74. doi: 10.4062/biomolther.2015.214. Biomol Ther (Seoul). 2016. PMID: 27169821 Free PMC article. Review.
Cited by
-
Exercise mimetics: a novel strategy to combat neuroinflammation and Alzheimer's disease.J Neuroinflammation. 2024 Feb 2;21(1):40. doi: 10.1186/s12974-024-03031-9. J Neuroinflammation. 2024. PMID: 38308368 Free PMC article. Review.
-
Tubular cell phenotype in HIV-associated nephropathy: role of phospholipid lysophosphatidic acid.Exp Mol Pathol. 2015 Aug;99(1):109-15. doi: 10.1016/j.yexmp.2015.06.004. Epub 2015 Jun 14. Exp Mol Pathol. 2015. PMID: 26079546 Free PMC article.
-
Autophagy of mitochondria: a promising therapeutic target for neurodegenerative disease.Cell Biochem Biophys. 2014 Nov;70(2):707-19. doi: 10.1007/s12013-014-0006-5. Cell Biochem Biophys. 2014. PMID: 24807843 Free PMC article. Review.
-
Roles of AMP-activated protein kinase in Alzheimer's disease.Neuromolecular Med. 2012 Mar;14(1):1-14. doi: 10.1007/s12017-012-8173-2. Epub 2012 Feb 26. Neuromolecular Med. 2012. PMID: 22367557 Review.
-
Endurance factors improve hippocampal neurogenesis and spatial memory in mice.Learn Mem. 2011 Jan 18;18(2):103-7. doi: 10.1101/lm.2001611. Print 2011 Feb. Learn Mem. 2011. PMID: 21245211 Free PMC article.
References
-
- Selkoe DJ. Alzheimer's disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis. 2001;3:75–80. - PubMed
-
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/S0197-4580(00)00124-X. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
