

The B1-subunit of the H(+) ATPase is required for maximal urinary acidification
- PMID: 16174750
- PMCID: PMC1224669
- DOI: 10.1073/pnas.0506769102
The B1-subunit of the H(+) ATPase is required for maximal urinary acidification
Abstract
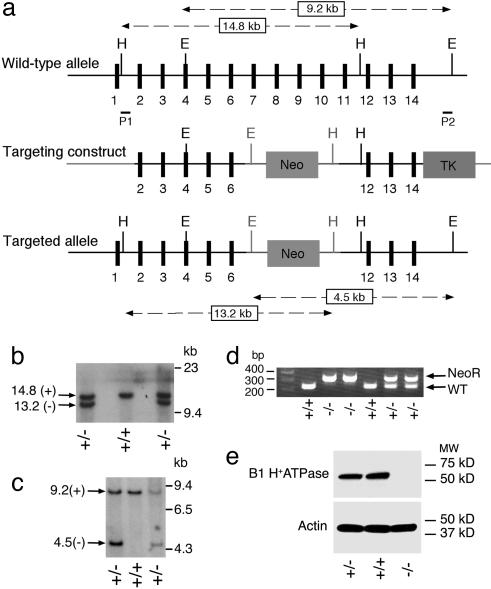
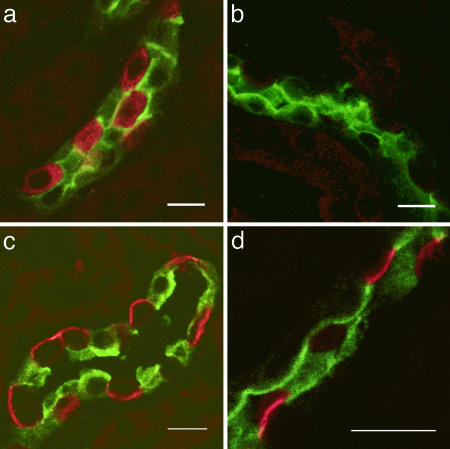
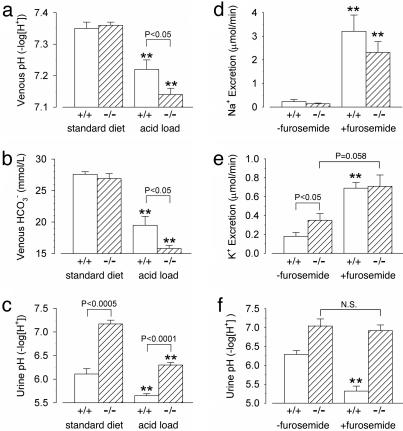
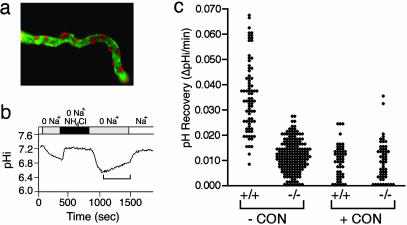
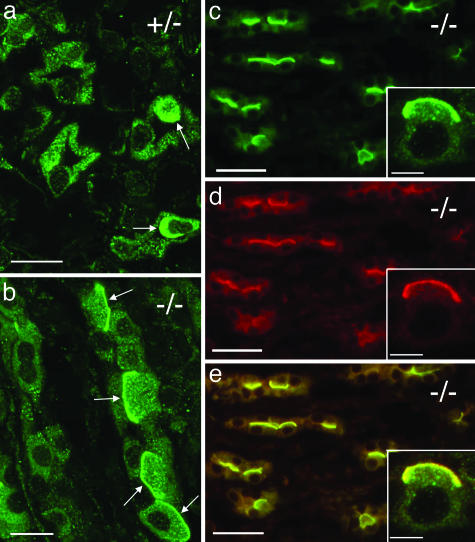
The multisubunit vacuolar-type H(+)ATPases mediate acidification of various intracellular organelles and in some tissues mediate H(+) secretion across the plasma membrane. Mutations in the B1-subunit of the apical H(+)ATPase that secretes protons in the distal nephron cause distal renal tubular acidosis in humans, a condition characterized by metabolic acidosis with an inappropriately alkaline urine. To examine the detailed cellular and organismal physiology resulting from this mutation, we have generated mice deficient in the B1-subunit (Atp6v1b1(-/-) mice). Urine pH is more alkaline and metabolic acidosis is more severe in Atp6v1b1(-/-) mice after oral acid challenge, demonstrating a failure of normal urinary acidification. In Atp6v1b1(-/-) mice, the normal urinary acidification induced by a lumen-negative potential in response to furosemide infusion is abolished. After an acute intracellular acidification, Na(+)-independent pH recovery rates of individual Atp6v1b1(-/-) intercalated cells of the cortical collecting duct are markedly reduced and show no further decrease after treatment with the selective H(+)ATPase inhibitor concanamycin. Apical expression of the alternative B-subunit isoform, B2, is increased in Atp6v1b1(-/-) medulla and colocalizes with the H(+)ATPase E-subunit; however, the greater severity of metabolic acidosis in Atp6v1b1(-/-) mice after oral acid challenge indicates that the B2-subunit cannot fully functionally compensate for the loss of B1. Our results indicate that the B1 isoform is the major B-subunit isoform that incorporates into functional, plasma membrane H(+)ATPases in intercalated cells of the cortical collecting duct and is required for maximal urinary acidification.
Figures
Similar articles
-
The B1 H + -ATPase ( Atp6v1b1 ) Subunit in Non-Type A Intercalated Cells is Required for Driving Pendrin Activity and the Renal Defense Against Alkalosis.J Am Soc Nephrol. 2024 Jan 1;35(1):7-21. doi: 10.1681/ASN.0000000000000259. Epub 2023 Nov 22. J Am Soc Nephrol. 2024. PMID: 37990364 Free PMC article.
-
The ammonia transporter RhCG modulates urinary acidification by interacting with the vacuolar proton-ATPases in renal intercalated cells.Kidney Int. 2018 Feb;93(2):390-402. doi: 10.1016/j.kint.2017.07.027. Epub 2017 Oct 18. Kidney Int. 2018. PMID: 29054531 Free PMC article.
-
Molecular cloning and characterization of Atp6v1b1, the murine vacuolar H+ -ATPase B1-subunit.Gene. 2003 Oct 30;318:25-34. doi: 10.1016/s0378-1119(03)00790-x. Gene. 2003. PMID: 14585495
-
Renal vacuolar H+-ATPase.Physiol Rev. 2004 Oct;84(4):1263-314. doi: 10.1152/physrev.00045.2003. Physiol Rev. 2004. PMID: 15383652 Review.
-
Regulation of luminal acidification by the V-ATPase.Physiology (Bethesda). 2013 Sep;28(5):318-29. doi: 10.1152/physiol.00007.2013. Physiology (Bethesda). 2013. PMID: 23997191 Free PMC article. Review.
Cited by
-
Altered V-ATPase expression in renal intercalated cells isolated from B1 subunit-deficient mice by fluorescence-activated cell sorting.Am J Physiol Renal Physiol. 2013 Mar 1;304(5):F522-32. doi: 10.1152/ajprenal.00394.2012. Epub 2012 Dec 26. Am J Physiol Renal Physiol. 2013. PMID: 23269648 Free PMC article.
-
Pathophysiology, diagnosis and treatment of inherited distal renal tubular acidosis.J Nephrol. 2018 Aug;31(4):511-522. doi: 10.1007/s40620-017-0447-1. Epub 2017 Oct 9. J Nephrol. 2018. PMID: 28994037 Review.
-
Extracellular pH regulates excitability of vomeronasal sensory neurons.J Neurosci. 2015 Mar 4;35(9):4025-39. doi: 10.1523/JNEUROSCI.2593-14.2015. J Neurosci. 2015. PMID: 25740530 Free PMC article.
-
Angiotensin II stimulates H⁺-ATPase activity in intercalated cells from isolated mouse connecting tubules and cortical collecting ducts.Cell Physiol Biochem. 2011;28(3):513-20. doi: 10.1159/000335112. Epub 2011 Nov 18. Cell Physiol Biochem. 2011. PMID: 22116365 Free PMC article.
-
Extra-renal locations of the a4 subunit of H(+)ATPase.BMC Cell Biol. 2016 Jul 2;17(1):27. doi: 10.1186/s12860-016-0106-8. BMC Cell Biol. 2016. PMID: 27368196 Free PMC article.
References
-
- Gennari, F. J. & Maddox, D. A. (2000) in The Kidney: Physiology and Pathophysiology, eds. Seldin, D. W. & Giebisch, G. (Lippincott, Philadelphia), pp. 2015-2053.
-
- DuBose, T. D. & Alpern, R. J. (2001) in The Metabolic & Molecular Bases of Inherited Disease, eds. Scriver, C. R., Beaudet, A. L., Sly, W. S. & Valle, D. (McGraw-Hill, New York), pp. 4983-5021.
-
- Hamm, L. L. & Alpern, R. J. (2000) in The Kidney: Physiology and Pathophysiology, eds. Seldin, D. W. & Giebisch, G. (Lippincott, Philadelphia), pp. 1935-1979.
-
- Karet, F. E., Finberg, K. E., Nelson, R. D., Nayir, A., Mocan, H., Sanjad, S. A., Rodriguez-Soriano, J., Santos, F., Cremers, C. W., Di Pietro, A., et al. (1999) Nat. Genet. 21, 84-90. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
