

PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway
- PMID: 16176978
- PMCID: PMC1289396
- DOI: 10.1091/mbc.e05-03-0268
PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway
Abstract
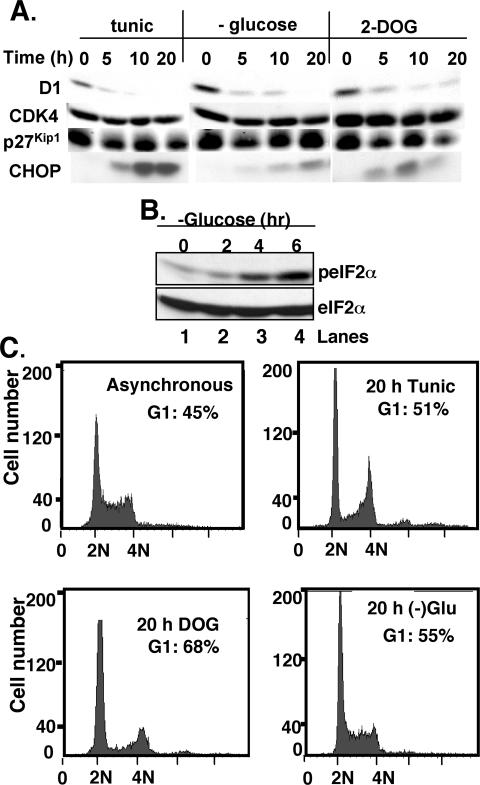
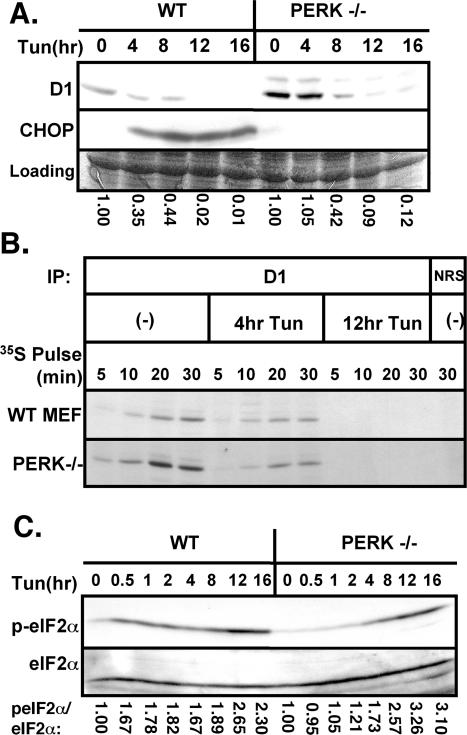
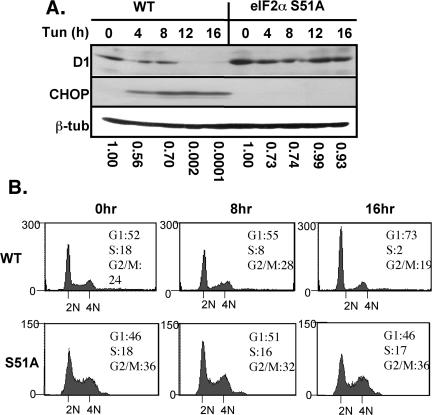
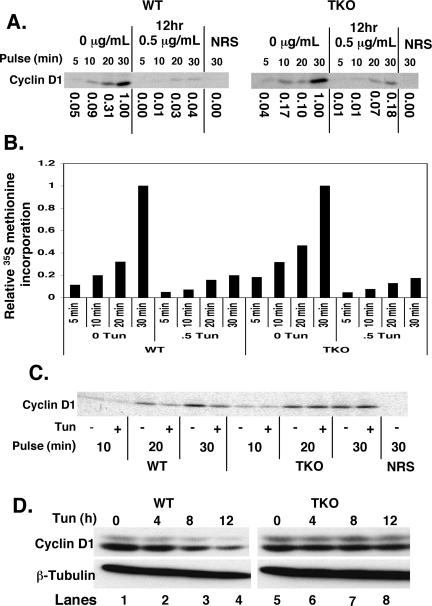
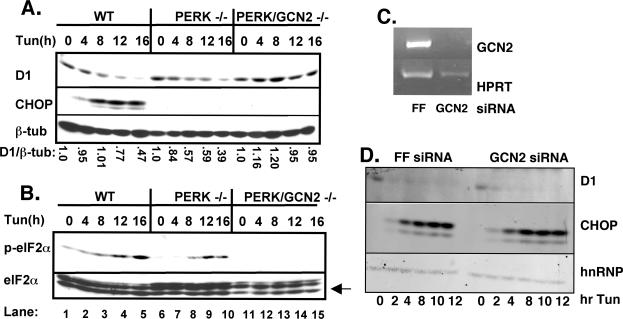
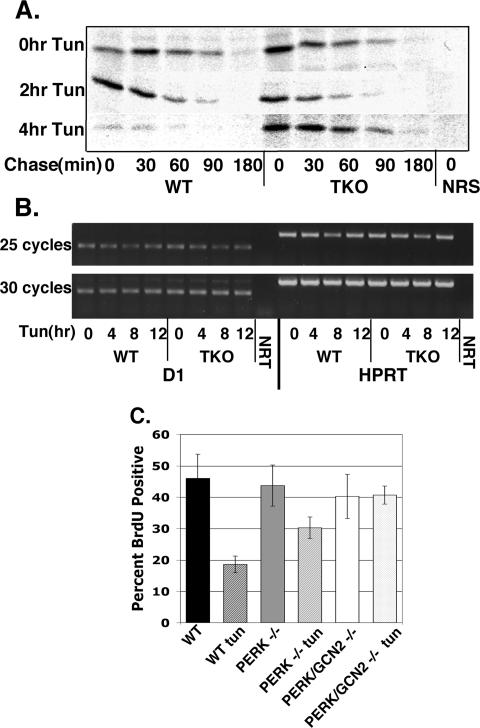
Exposure of cells to endoplasmic reticulum (ER) stress leads to activation of PKR-like ER kinase (PERK), eukaryotic translation initiation factor 2alpha (eIF2alpha) phosphorylation, repression of cyclin D1 translation, and subsequent cell cycle arrest in G1 phase. However, whether PERK is solely responsible for regulating cyclin D1 accumulation after unfolded protein response pathway (UPR) activation has not been assessed. Herein, we demonstrate that repression of cyclin D1 translation after UPR activation occurs independently of PERK, but it remains dependent on eIF2alpha phosphorylation. Although phosphorylation of eIF2alpha in PERK-/- fibroblasts is attenuated in comparison with wild-type fibroblasts, it is not eliminated. The residual eIF2alpha phosphorylation correlates with the kinetics of cyclin D1 loss, suggesting that another eIF2alpha kinase functions in the absence of PERK. In cells harboring targeted deletion of both PERK and GCN2, cyclin D1 loss is attenuated, suggesting GCN2 functions as the redundant kinase. Consistent with these results, cyclin D1 translation is also stabilized in cells expressing a nonphosphorylatable allele of eIF2alpha; in contrast, repression of global protein translation still occurs in these cells, highlighting a high degree of specificity in transcripts targeted for translation inhibition by phosphorylated eIF2alpha. Our results demonstrate that PERK and GCN2 function to cooperatively regulate eIF2alpha phosphorylation and cyclin D1 translation after UPR activation.
Figures
References
-
- Cox, J. S., Shamu, C. E., and Walter, P. (1993). Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206. - PubMed
-
- Cullinan, S. B., and Diehl, J. A. (2004). PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following ER stress. J. Biol. Chem. 279, 20076–20087. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
