

Microarray based comparative genomic hybridization testing in deletion bearing patients with Angelman syndrome: genotype-phenotype correlations
- PMID: 16183798
- PMCID: PMC2564536
- DOI: 10.1136/jmg.2005.036913
Microarray based comparative genomic hybridization testing in deletion bearing patients with Angelman syndrome: genotype-phenotype correlations
Abstract
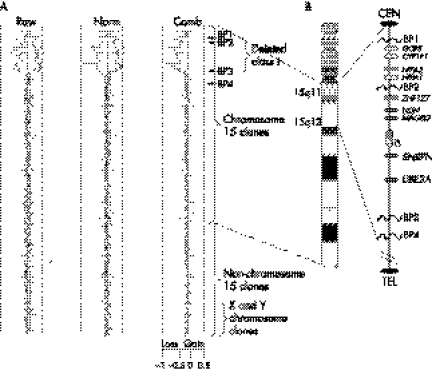
Background: Angelman syndrome (AS) is a neurodevelopmental disorder characterised by severe mental retardation, dysmorphic features, ataxia, seizures, and typical behavioural characteristics, including a happy sociable disposition. AS is caused by maternal deficiency of UBE3A (E6 associated protein ubiquitin protein ligase 3A gene), located in an imprinted region on chromosome 15q11-q13. Although there are four different molecular types of AS, deletions of the 15q11-q13 region account for approximately 70% of the AS patients. These deletions are usually detected by fluorescence in situ hybridisation studies. The deletions can also be subclassified based on their size into class I and class II, with the former being larger and encompassing the latter.
Methods: We studied 22 patients with AS due to microdeletions using a microarray based comparative genomic hybridisation (array CGH) assay to define the deletions and analysed their phenotypic severity, especially expression of the autism phenotype, in order to establish clinical correlations.
Results: Overall, children with larger, class I deletions were significantly more likely to meet criteria for autism, had lower cognitive scores, and lower expressive language scores compared with children with smaller, class II deletions. Children with class I deletions also required more medications to control their seizures than did those in the class II group.
Conclusions: There are four known genes (NIPA1, NIPA2, CYFIP1, & GCP5) that are affected by class I but not class II deletions, thus raising the possibility of a role for these genes in autism as well as the development of expressive language skills.
Conflict of interest statement
Competing interests: there are no competing interests
Similar articles
-
Neurodevelopmental outcome in Angelman syndrome: genotype-phenotype correlations.Res Dev Disabil. 2014 Jul;35(7):1742-7. doi: 10.1016/j.ridd.2014.02.018. Epub 2014 Mar 19. Res Dev Disabil. 2014. PMID: 24656292
-
Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations.Eur J Hum Genet. 2007 Sep;15(9):943-9. doi: 10.1038/sj.ejhg.5201859. Epub 2007 May 23. Eur J Hum Genet. 2007. PMID: 17522620
-
Distinct phenotypes distinguish the molecular classes of Angelman syndrome.J Med Genet. 2001 Dec;38(12):834-45. doi: 10.1136/jmg.38.12.834. J Med Genet. 2001. PMID: 11748306 Free PMC article.
-
Genotype-Phenotype Correlations in Angelman Syndrome.Genes (Basel). 2021 Jun 28;12(7):987. doi: 10.3390/genes12070987. Genes (Basel). 2021. PMID: 34203304 Free PMC article. Review.
-
Novel intragenic deletions within the UBE3A gene in two unrelated patients with Angelman syndrome: case report and review of the literature.BMC Med Genet. 2017 Nov 21;18(1):137. doi: 10.1186/s12881-017-0500-x. BMC Med Genet. 2017. PMID: 29162042 Free PMC article. Review.
Cited by
-
Laryngotracheal separation surgery in a patient with severe Angelman syndrome involving a 19.3 Mb deletion on 15q11.2-q14.Clin Case Rep. 2022 Nov 6;10(11):e6545. doi: 10.1002/ccr3.6545. eCollection 2022 Nov. Clin Case Rep. 2022. PMID: 36381038 Free PMC article.
-
Regulation of molecular pathways in the Fragile X Syndrome: insights into Autism Spectrum Disorders.J Neurodev Disord. 2011 Sep;3(3):257-69. doi: 10.1007/s11689-011-9087-2. Epub 2011 Aug 13. J Neurodev Disord. 2011. PMID: 21842222 Free PMC article.
-
Autism Spectrum Disorder Symptom Profiles in Fragile X Syndrome, Angelman Syndrome, Tuberous Sclerosis Complex and Neurofibromatosis Type 1.J Autism Dev Disord. 2024 Oct 12. doi: 10.1007/s10803-024-06557-2. Online ahead of print. J Autism Dev Disord. 2024. PMID: 39395123
-
Epilepsy and Molecular Phenotype Affect the Neurodevelopment of Pediatric Angelman Syndrome Patients in China.Front Psychiatry. 2022 Apr 28;13:886028. doi: 10.3389/fpsyt.2022.886028. eCollection 2022. Front Psychiatry. 2022. PMID: 35573374 Free PMC article.
-
Genomic analysis of the chromosome 15q11-q13 Prader-Willi syndrome region and characterization of transcripts for GOLGA8E and WHCD1L1 from the proximal breakpoint region.BMC Genomics. 2008 Jan 28;9:50. doi: 10.1186/1471-2164-9-50. BMC Genomics. 2008. PMID: 18226259 Free PMC article.
References
-
- Angelman H. “Puppet children”: a report of three cases. Dev Med Child Neurol 19657681–688. - PubMed
-
- Berg J M, Pakula Z. Angelman's (“happy puppet”) syndrome. Am J Dis Child 197212372–74. - PubMed
-
- Williams C A, Angelman H, Clayton‐Smith J, Driscoll D J, Hendrickson J E, Knoll J H, Magenis R E, Schinzel A, Wagstaff J, Whidden E M, Zori R T. Angelman syndrome: consensus for diagnostic criteria. Angelman Syndrome Foundation. Am J Med Genet 199556237–238. - PubMed
-
- Fryburg J S, Breg W R, Lindgren V. Diagnosis of Angelman syndrome in infants. Am J Med Genet 19913858–64. - PubMed
-
- Magenis R E, Brown M G, Lacy D A, Budden S, LaFranchi S. Is Angelman syndrome an alternate result of del(15)(q11q13)? Am J Med Genet 198728829–838. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials