

ETHE1 mutations are specific to ethylmalonic encephalopathy
- PMID: 16183799
- PMCID: PMC2563233
- DOI: 10.1136/jmg.2005.036210
ETHE1 mutations are specific to ethylmalonic encephalopathy
Abstract
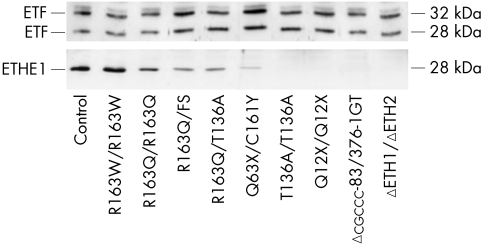
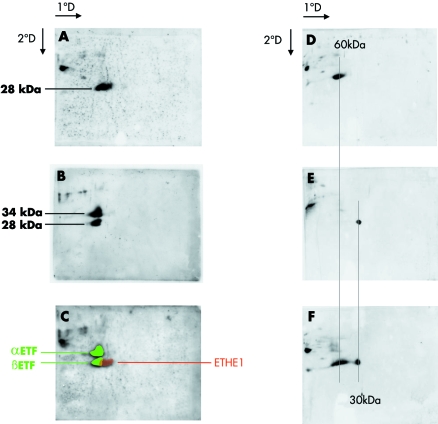
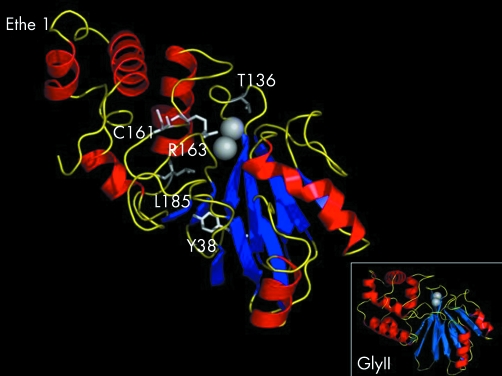
Mutations in ETHE1, a gene located at chromosome 19q13, have recently been identified in patients affected by ethylmalonic encephalopathy (EE). EE is a devastating infantile metabolic disorder, characterised by widespread lesions in the brain, hyperlactic acidaemia, petechiae, orthostatic acrocyanosis, and high levels of ethylmalonic acid in body fluids. To investigate to what extent ETHE1 is responsible for EE, we analysed this gene in 29 patients with typical EE and in 11 patients presenting with early onset progressive encephalopathy with ethylmalonic aciduria (non-EE EMA). Frameshift, stop, splice site, and missense mutations of ETHE1 were detected in all the typical EE patients analysed. Western blot analysis of the ETHE1 protein indicated that some of the missense mutations are associated with the presence of the protein, suggesting that the corresponding wild type amino acid residues have a catalytic function. No ETHE1 mutations were identified in non-EE EMA patients. Experiments based on two dimensional blue native electrophoresis indicated that ETHE1 protein works as a supramolecular, presumably homodimeric, complex, and a three dimensional model of the protein suggests that it is likely to be a mitochondrial matrix thioesterase acting on a still unknown substrate. Finally, the 625G-->A single nucleotide polymorphism in the gene encoding the short chain acyl-coenzyme A dehydrogenase (SCAD) was previously proposed as a co-factor in the aetiology of EE and other EMA syndromes. SNP analysis in our patients ruled out a pathogenic role of SCAD variants in EE, but did show a highly significant prevalence of the 625A alleles in non-EE EMA patients.
Conflict of interest statement
Competing interests: there are no competing interests.
References
-
- Burlina A, Zacchello F, Dionisi‐Vici C, Bertini E, Sabetta G, Bennet M J, Hale D E, Schmidt‐Sommerfeld E, Rinaldo P. New clinical phenotype of branched‐chain acyl‐CoA oxidation defect. Lancet 1991141522–1523. - PubMed
-
- Grosso S, Balestri P, Mostardini R, Federico A, De Stefano N. Brain mitochondrial impairment in ethylmalonic encephalopathy. J Neurol 2004251755–756. - PubMed
-
- Tiranti V, D'Adamo P, Briem E, Ferrari G, Mineri R, Lamantea E, Mandel H, Balestri P, Garcia‐Silva M T, Vollmer B, Rinaldo P, Hahn S H, Leonard J, Rahman S, Dionisi‐Vici C, Garavaglia B, Gasparini P, Zeviani M. Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am J Hum Genet 200474239–252. - PMC - PubMed
-
- Garcia‐Silva M T, Ribes A, Campos Y, Garavaglia B, Arenas J. Syndrome of encephalopathy, petechiae, and ethylmalonic aciduria. Pediatr Neurol 199717165–170. - PubMed
-
- Nowaczyk M J, Lehotay D C, Platt B A, Fisher L, Tan R, Phillips H, Clarke J T. Ethylmalonic and methylsuccinic aciduria in ethylmalonic encephalopathy arise from abnormal isoleucine metabolism. Metabolism 199847836–839. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials