

Ablation of oncogenic ALK is a viable therapeutic approach for anaplastic large-cell lymphomas
- PMID: 16189272
- PMCID: PMC1895619
- DOI: 10.1182/blood-2005-05-2125
Ablation of oncogenic ALK is a viable therapeutic approach for anaplastic large-cell lymphomas
Abstract
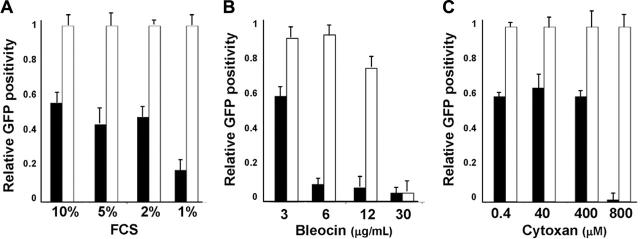
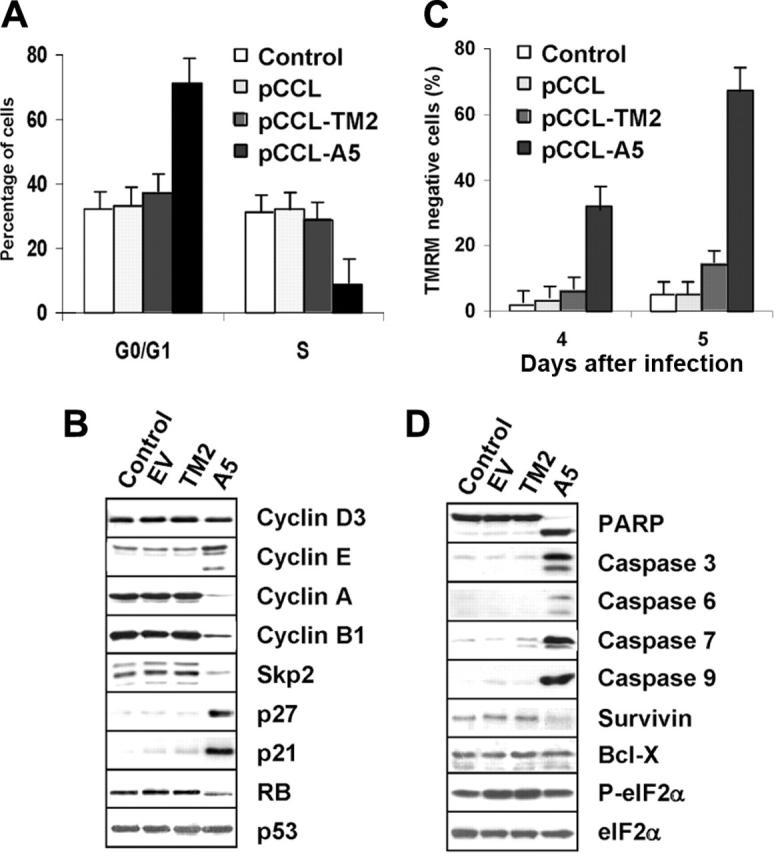
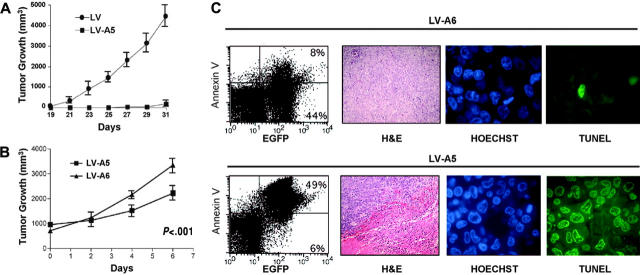
Anaplastic large-cell lymphomas (ALCLs) carry chromosome translocations in which the anaplastic lymphoma kinase (ALK) gene is fused to several partners, most frequently, the NPM1 gene. We have demonstrated that the constitutive activation of ALK fusion proteins results in cellular transformation and lymphoid neoplasia. Herein, we specifically down-regulated ALK protein expression by using small hairpin RNA (shRNA) targeting a sequence coding for the catalytic domain of ALK. The ablation of ALK leads to the down-modulation of known ALK downstream effectors, cell growth arrest, and reversion of the transformed phenotype of ALK(+) mouse embryonic fibroblasts in vitro and in vivo. In human ALCL cells lentiviral-mediated ALK knock-down leads to G(1) cell-cycle arrest and apoptosis in vitro and tumor growth inhibition and regression in vivo. Using a specific approach we have demonstrated that the survival and growth of ALK(+) ALCLs are strictly dependent on ALK activation and signaling. Therefore, ALK is a viable target for therapeutic intervention and its inactivation might represent a pivotal approach for the treatment of ALK lymphomas and other ALK-dependent human tumors.
Figures




References
-
- Stein H, Foss HD, Durkop H, et al. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000;96: 3681-3695. - PubMed
-
- Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263: 1281-1284. - PubMed
-
- Duyster J, Bai RY, Morris SW. Translocations involving anaplastic lymphoma kinase (ALK). Oncogene. 2001;20: 5623-5637. - PubMed
-
- Cessna MH, Zhou H, Sanger WG, et al. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol. 2002;15: 931-938. - PubMed
-
- Onciu M, Behm FG, Downing JR, et al. ALK-positive plasmablastic B-cell lymphoma with expression of the NPM-ALK fusion transcript: report of 2 cases. Blood. 2003;102: 2642-2644. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
