

Deregulation of the phosphatidylinositol-3 kinase signaling cascade is associated with neurodegeneration in Npc1-/- mouse brain
- PMID: 16192643
- PMCID: PMC1603683
- DOI: 10.1016/S0002-9440(10)61197-2
Deregulation of the phosphatidylinositol-3 kinase signaling cascade is associated with neurodegeneration in Npc1-/- mouse brain
Abstract
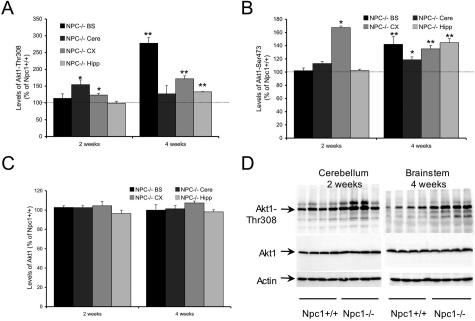
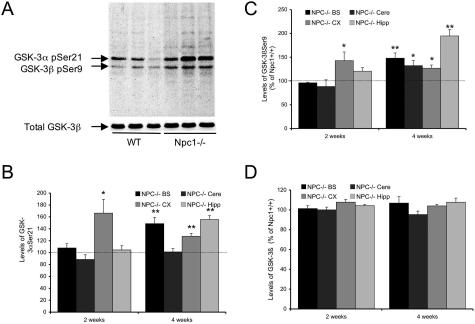
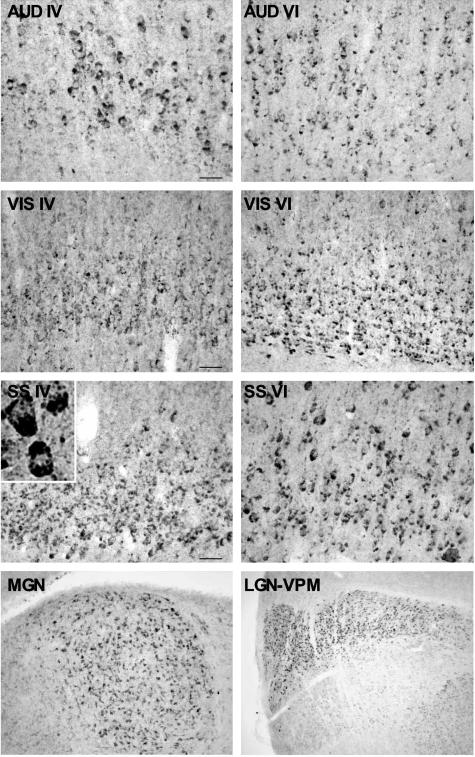
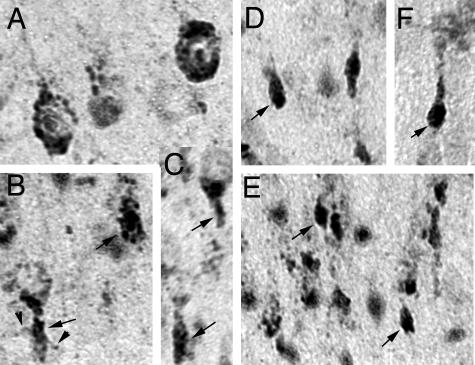
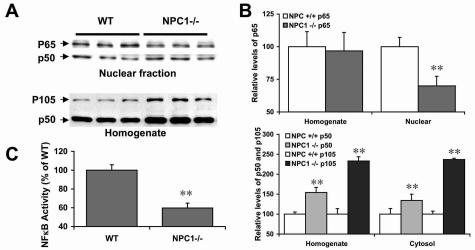
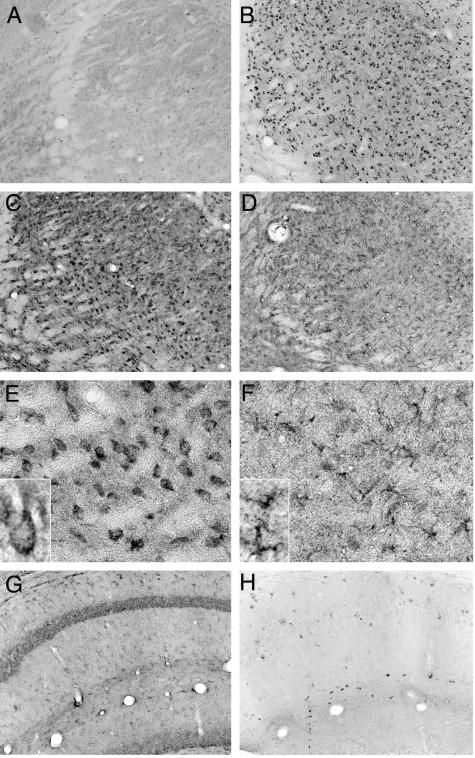
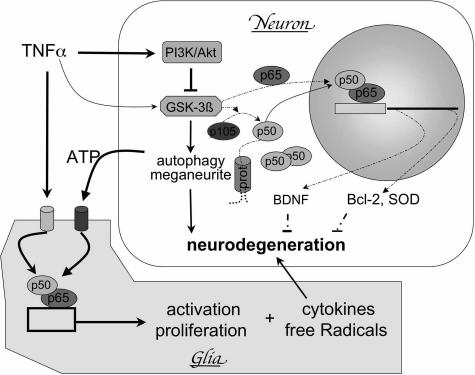
Niemann-Pick type C (NPC) disease is caused by mutations to genes that encode proteins critical to intracellular lipid homeostasis. The events underlying NPC progressive neurodegeneration are poorly understood but include neurofibrillary tangles of the type found in Alzheimer's disease. Here we investigated possible contributions of a phosphatidylinositol-3 kinase cascade [PI3K, Akt, glycogen synthase kinase-3beta (GSK-3beta)] that is linked to apoptosis and various degenerative conditions. Brain concentrations of phosphorylated Akt, which phosphorylates and inactivates GSK-3beta, were significantly elevated in Npc1-/- mice relative to Npc1+/+ mice. Accordingly, levels of inactive GSK-3beta were 50 to 100% higher in mutant brains than in controls. Increases in inactive GSK-3beta occurred early in postnatal development, well before neuronal loss, and were most prominent in structures with intracellular cholesterol accumulation, suggesting a contribution to subsequent degeneration. Perturbations of nuclear factor (NF)-kappaB, which is regulated by GSK-3beta, occurred in Npc1-/- mouse brains. Nuclear concentrations and DNA binding activity of NF-kappaB's transactivation subunit, p65, were significantly reduced in Npc1-/- mice compared to Npc1+/+ mice. Cytoplasmic levels of the p50 subunit and its precursor, p105, were higher in Npc1-/- mice. These results suggest that excessive activity in the PI3K-Akt pathway depresses GSK-3beta, thereby disrupting the formation and/or nuclear import of p50/p65 NF-kappaB dimers and contributing to neuronal degeneration.
Figures
Similar articles
-
Neurodegeneration in heterozygous Niemann-Pick type C1 (NPC1) mouse: implication of heterozygous NPC1 mutations being a risk for tauopathy.J Biol Chem. 2005 Jul 22;280(29):27296-302. doi: 10.1074/jbc.M503922200. Epub 2005 May 25. J Biol Chem. 2005. PMID: 15919659
-
Site-specific phosphorylation of tau accompanied by activation of mitogen-activated protein kinase (MAPK) in brains of Niemann-Pick type C mice.J Biol Chem. 2001 Mar 30;276(13):10314-9. doi: 10.1074/jbc.M009733200. Epub 2001 Jan 4. J Biol Chem. 2001. PMID: 11152466
-
Glycogen synthase kinase 3beta functions to specify gene-specific, NF-kappaB-dependent transcription.Mol Cell Biol. 2005 Oct;25(19):8444-55. doi: 10.1128/MCB.25.19.8444-8455.2005. Mol Cell Biol. 2005. PMID: 16166627 Free PMC article.
-
GSK-3β at the Intersection of Neuronal Plasticity and Neurodegeneration.Neural Plast. 2019 May 2;2019:4209475. doi: 10.1155/2019/4209475. eCollection 2019. Neural Plast. 2019. PMID: 31191636 Free PMC article. Review.
-
Primers on molecular pathways. The glycogen synthase kinase-3beta.Pancreatology. 2007;7(5-6):398-402. doi: 10.1159/000108955. Epub 2007 Oct 1. Pancreatology. 2007. PMID: 17912008 Free PMC article. Review.
Cited by
-
Increased activity and altered subcellular distribution of lysosomal enzymes determine neuronal vulnerability in Niemann-Pick type C1-deficient mice.Am J Pathol. 2009 Dec;175(6):2540-56. doi: 10.2353/ajpath.2009.081096. Epub 2009 Nov 5. Am J Pathol. 2009. PMID: 19893049 Free PMC article.
-
Perturbed rhythmic activation of signaling pathways in mice deficient for Sterol Carrier Protein 2-dependent diurnal lipid transport and metabolism.Sci Rep. 2016 Apr 21;6:24631. doi: 10.1038/srep24631. Sci Rep. 2016. PMID: 27097688 Free PMC article.
-
Endosomal accumulation of Toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal transducers and activators of transcription in Niemann-Pick disease type C (NPC) fibroblasts: a potential basis for glial cell activation in the NPC brain.J Neurosci. 2007 Feb 21;27(8):1879-91. doi: 10.1523/JNEUROSCI.5282-06.2007. J Neurosci. 2007. PMID: 17314284 Free PMC article.
-
Altered levels and distribution of amyloid precursor protein and its processing enzymes in Niemann-Pick type C1-deficient mouse brains.Glia. 2010 Aug 15;58(11):1267-81. doi: 10.1002/glia.21001. Glia. 2010. PMID: 20607864 Free PMC article.
-
Novel mechanism of U18666A-induced tumour necrosis factor-alpha production in RAW 264.7 macrophage cells.Clin Exp Immunol. 2009 Mar;155(3):552-8. doi: 10.1111/j.1365-2249.2008.03779.x. Clin Exp Immunol. 2009. PMID: 19220841 Free PMC article.
References
-
- Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie EJ, Dwyer NK, Neufeld EB, Chang TY, Liscum L, Strauss JF, 3rd, Ohno K, Zeigler M, Carmi R, Sokol J, Markie D, O’Neill RR, van Diggelen OP, Elleder M, Patterson MC, Brady RO, Vanier MT, Pentchev PG, Tagle DA. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. - PubMed
-
- Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–2301. - PubMed
-
- Auer IA, Schmidt ML, Lee VM, Curry B, Suzuki K, Shin RW, Pentchev PG, Carstea ED, Trojanowski JQ. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer’s disease. Acta Neuropathol (Berl) 1995;90:547–551. - PubMed
-
- Suzuki K, Parker CC, Pentchev PG, Katz D, Ghetti B, D’Agostino AN, Carstea ED. Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol (Berl) 1995;89:227–238. - PubMed
-
- Suzuki H, Sakiyama T, Harada N, Abe M, Tadokoro M. Pathologic changes of glial cells in murine model of Niemann-Pick disease type C: immunohistochemical, lectin-histochemical and ultrastructural observations. Pediatr Int. 2003;45:1–4. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
