

Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1
- PMID: 16201016
- PMCID: PMC1238739
- DOI: 10.1371/journal.ppat.0010004
Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1
Abstract
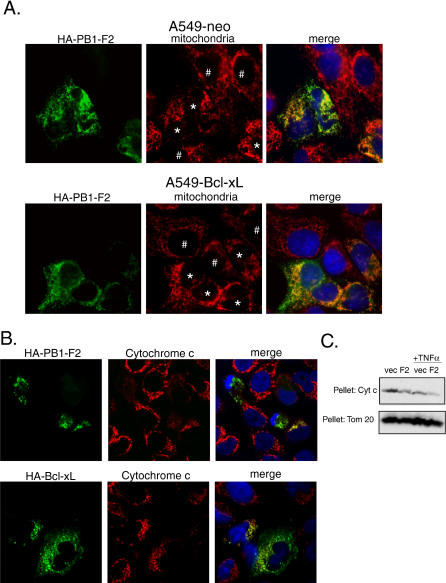
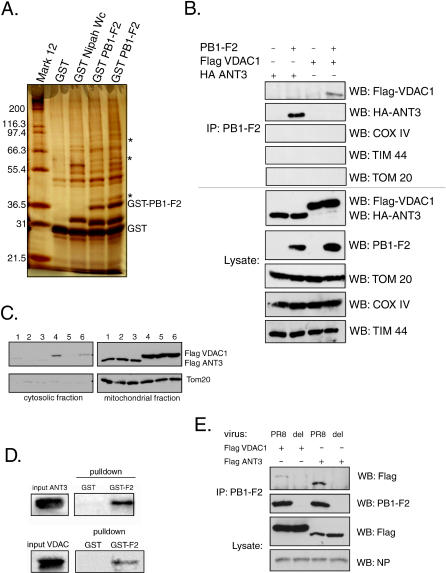
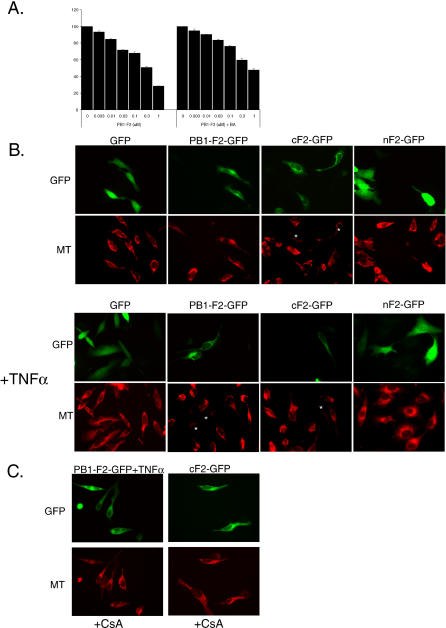
The influenza virus PB1-F2 is an 87-amino acid mitochondrial protein that previously has been shown to induce cell death, although the mechanism of apoptosis induction has remained unclear. In the process of characterizing its mechanism of action we found that the viral PB1-F2 protein sensitizes cells to apoptotic stimuli such as tumor necrosis factor alpha, as demonstrated by increased cleavage of caspase 3 substrates in PB1-F2-expressing cells. Moreover, treatment of purified mouse liver mitochondria with recombinant PB1-F2 protein resulted in cytochrome c release, loss of the mitochondrial membrane potential, and enhancement of tBid-induced mitochondrial permeabilization, suggesting a possible mechanism for the observed cellular sensitization to apoptosis. Using glutathione-S-transferase pulldowns with subsequent mass spectrometric analysis, we identified the mitochondrial interactors of the PB1-F2 protein and showed that the viral protein uniquely interacts with the inner mitochondrial membrane adenine nucleotide translocator 3 and the outer mitochondrial membrane voltage-dependent anion channel 1, both of which are implicated in the mitochondrial permeability transition during apoptosis. Consistent with this interaction, blockers of the permeability transition pore complex (PTPC) inhibited PB1-F2-induced mitochondrial permeabilization. Based on our findings, we propose a model whereby the proapoptotic PB1-F2 protein acts through the mitochondrial PTPC and may play a role in the down-regulation of the host immune response to infection.
Conflict of interest statement
Figures





References
-
- Garcia-Sastre A. Inhibition of interferon-mediated antiviral responses by influenza A viruses and other negative-strand RNA viruses. Virology. 2001;279:375–384. - PubMed
-
- Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, et al. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–330. - PubMed
-
- Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med. 2001;7:1306–1312. - PubMed
-
- Yamada H, Chounan R, Higashi Y, Kurihara N, Kido H. Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 2004;578:331–336. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
