

Persistent NF-kappaB activation in renal epithelial cells in a mouse model of HIV-associated nephropathy
- PMID: 16204413
- PMCID: PMC1892240
- DOI: 10.1152/ajprenal.00208.2005
Persistent NF-kappaB activation in renal epithelial cells in a mouse model of HIV-associated nephropathy
Abstract
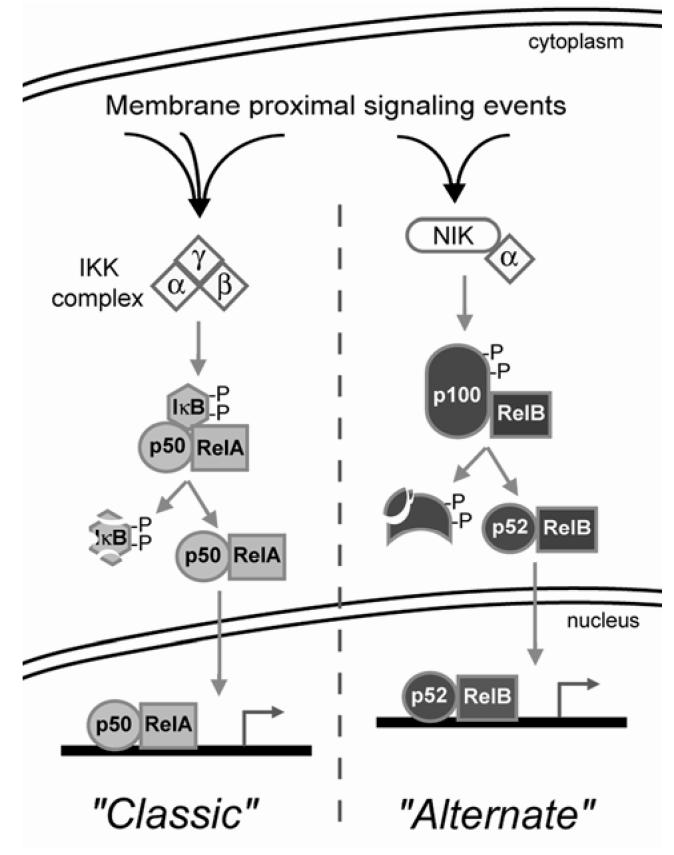
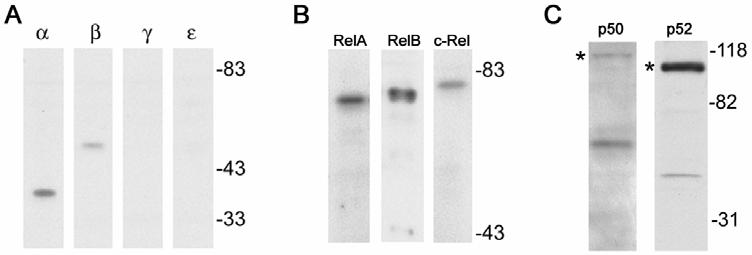
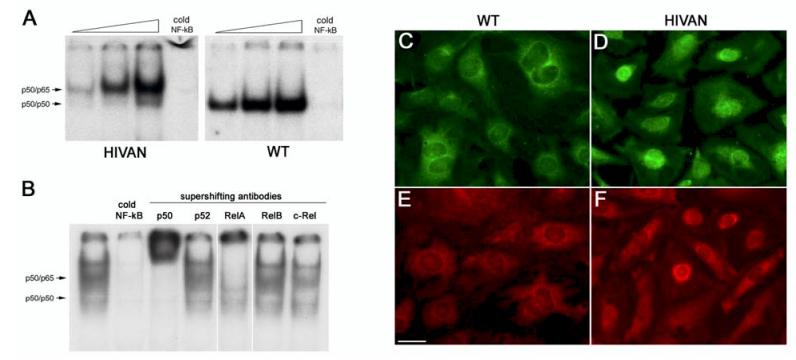
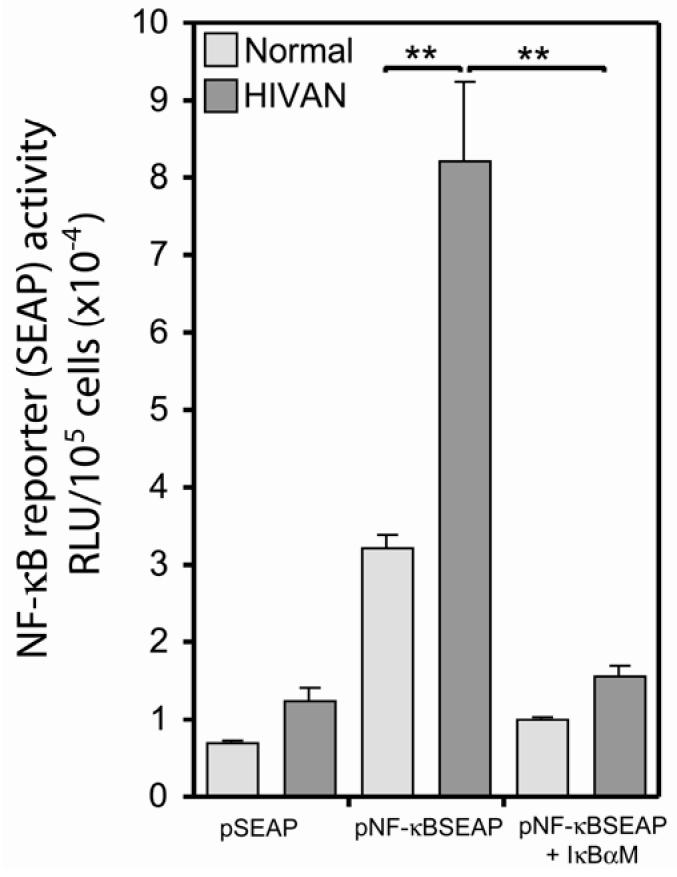
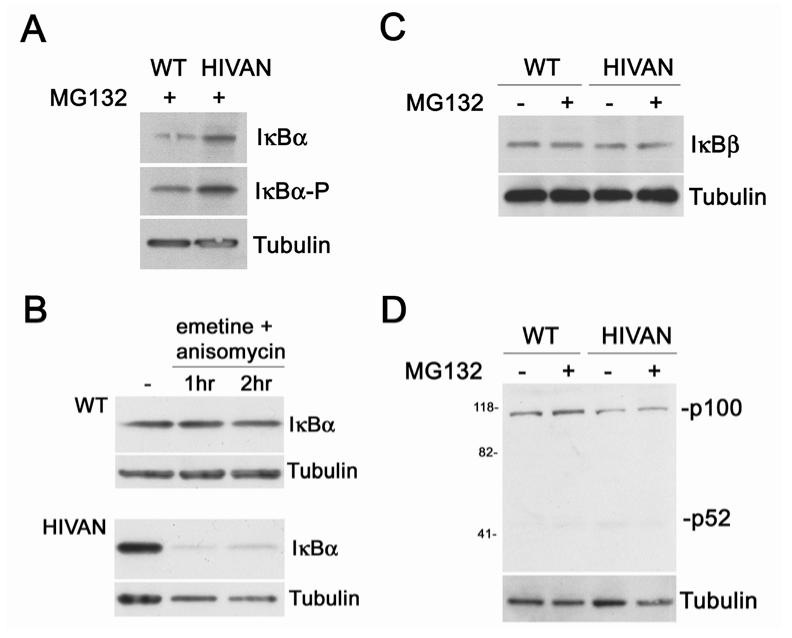
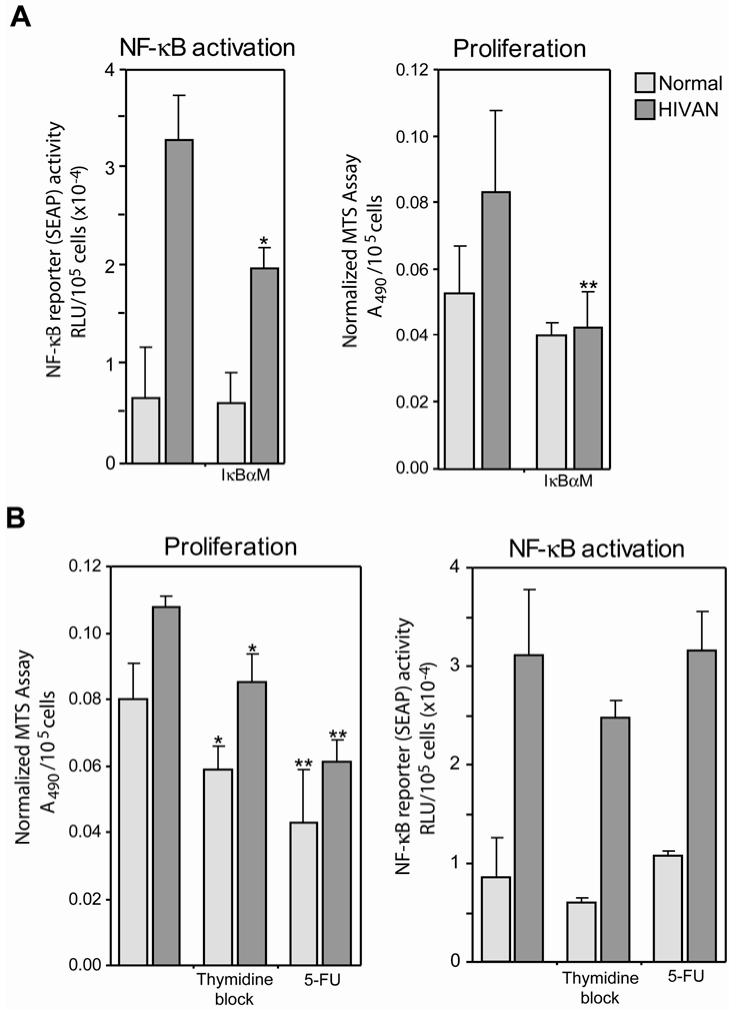
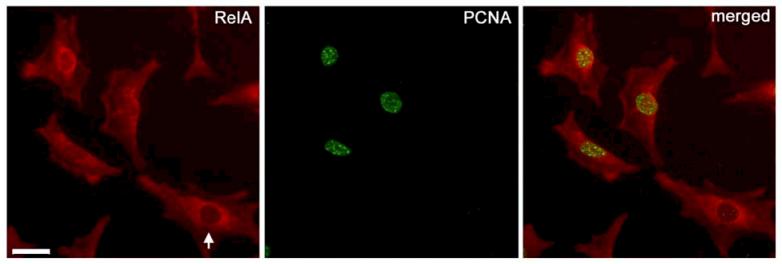
Human immunodeficiency virus (HIV)-associated nephropathy (HIVAN) is caused, in part, by direct infection of kidney epithelial cells by HIV-1. In the spectrum of pathogenic host-virus interactions, abnormal activation or suppression of host transcription factors is common. NF-kappaB is a necessary host transcription factor for HIV-1 gene expression, and it has been shown that NF-kappaB activity is dysregulated in many naturally infected cell types. We show here that renal glomerular epithelial cells (podocytes) expressing the HIV-1 genome, similar to infected immune cells, also have a dysregulated and persistent activation of NF-kappaB. Although podocytes produce p50, p52, RelA, RelB, and c-Rel, electrophoretic mobility shift assays and immunocytochemistry showed a predominant nuclear accumulation of p50/RelA-containing NF-kappaB dimers in HIV-1-expressing podocytes compared with normal. In addition, the expression level of a transfected NF-kappaB reporter plasmid was significantly higher in HIVAN podocytes. The mechanism of NF-kappaB activation involved increased phosphorylation of IkappaBalpha, resulting in an enhanced turnover of the IkappaBalpha protein. There was no evidence for regulation by IkappaBbeta or the alternate pathway of NF-kappaB activation. Altered activation of this key host transcription factor likely plays a role in the well-described cellular phenotypic changes observed in HIVAN, such as proliferation. Studies with inhibitors of proliferation and NF-kappaB suggest that NF-kappaB activation may contribute to the proliferative mechanism in HIVAN. In addition, because NF-kappaB regulates many aspects of inflammation, this dysregulation may also contribute to disease severity and progression through regulation of proinflammatory processes in the kidney microenvironment.
Figures
References
-
- Barisoni L, Kriz W, Mundel P, D'Agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 1999;10:51–61. - PubMed
-
- Barisoni L, Mokrzycki M, Sablay L, Nagata M, Yamase H, Mundel P. Podocyte cell cycle regulation and proliferation in collapsing glomerulopathies. Kidney Int. 2000;58:137–143. - PubMed
-
- Barisoni L, Mundel P. Podocyte biology and the emerging understanding of podocyte diseases. Am.J.Nephrol. 2003;23:353–360. - PubMed
-
- Bird JE, Durham SK, Giancarli MR, Gitlitz PH, Pandya DG, Dambach DM, Mozes MM, Kopp JB. Captopril prevents nephropathy in HIV-transgenic mice. J.Am.Soc.Nephrol. 1998;9:1441–1447. - PubMed
-
- Bruggeman LA, Adler SH, Klotman PE. Nuclear factor-kappa B binding to the HIV-1 LTR in kidney: implications for HIV-associated nephropathy. Kidney Int. 2001;59:2174–2181. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials