

ASPIC: a novel method to predict the exon-intron structure of a gene that is optimally compatible to a set of transcript sequences
- PMID: 16207377
- PMCID: PMC1276783
- DOI: 10.1186/1471-2105-6-244
ASPIC: a novel method to predict the exon-intron structure of a gene that is optimally compatible to a set of transcript sequences
Abstract
Background: Currently available methods to predict splice sites are mainly based on the independent and progressive alignment of transcript data (mostly ESTs) to the genomic sequence. Apart from often being computationally expensive, this approach is vulnerable to several problems--hence the need to develop novel strategies.
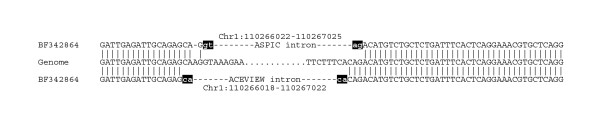
Results: We propose a method, based on a novel multiple genome-EST alignment algorithm, for the detection of splice sites. To avoid limitations of splice sites prediction (mainly, over-predictions) due to independent single EST alignments to the genomic sequence our approach performs a multiple alignment of transcript data to the genomic sequence based on the combined analysis of all available data. We recast the problem of predicting constitutive and alternative splicing as an optimization problem, where the optimal multiple transcript alignment minimizes the number of exons and hence of splice site observations. We have implemented a splice site predictor based on this algorithm in the software tool ASPIC (Alternative Splicing PredICtion). It is distinguished from other methods based on BLAST-like tools by the incorporation of entirely new ad hoc procedures for accurate and computationally efficient transcript alignment and adopts dynamic programming for the refinement of intron boundaries. ASPIC also provides the minimal set of non-mergeable transcript isoforms compatible with the detected splicing events. The ASPIC web resource is dynamically interconnected with the Ensembl and Unigene databases and also implements an upload facility.
Conclusion: Extensive bench marking shows that ASPIC outperforms other existing methods in the detection of novel splicing isoforms and in the minimization of over-predictions. ASPIC also requires a lower computation time for processing a single gene and an EST cluster. The ASPIC web resource is available at http://aspic.algo.disco.unimib.it/aspic-devel/.
Figures
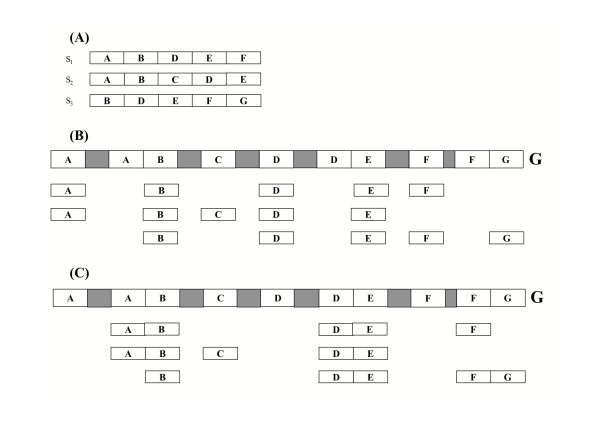
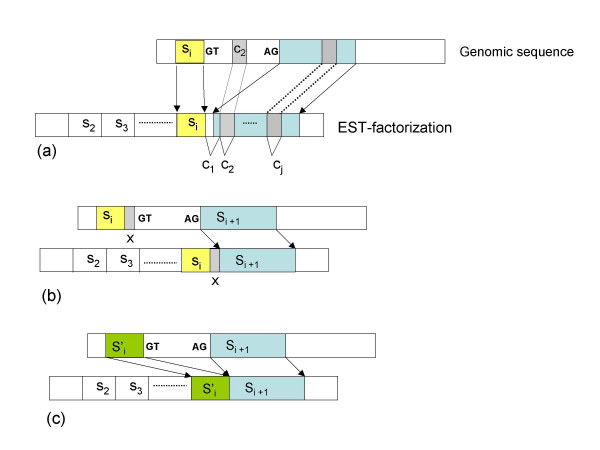
 inducing canonical splice sites on the genomic sequence can be optimally aligned before si+1 thus leading to an optimal location of both si and si+1.
inducing canonical splice sites on the genomic sequence can be optimally aligned before si+1 thus leading to an optimal location of both si and si+1.

Similar articles
-
ASPIC: a web resource for alternative splicing prediction and transcript isoforms characterization.Nucleic Acids Res. 2006 Jul 1;34(Web Server issue):W440-3. doi: 10.1093/nar/gkl324. Nucleic Acids Res. 2006. PMID: 16845044 Free PMC article.
-
PIntron: a fast method for detecting the gene structure due to alternative splicing via maximal pairings of a pattern and a text.BMC Bioinformatics. 2012 Apr 12;13 Suppl 5(Suppl 5):S2. doi: 10.1186/1471-2105-13-S5-S2. BMC Bioinformatics. 2012. PMID: 22537006 Free PMC article.
-
SplicedFamAlign: CDS-to-gene spliced alignment and identification of transcript orthology groups.BMC Bioinformatics. 2019 Mar 29;20(Suppl 3):133. doi: 10.1186/s12859-019-2647-2. BMC Bioinformatics. 2019. PMID: 30925859 Free PMC article.
-
Computational discovery of human coding and non-coding transcripts with conserved splice sites.Bioinformatics. 2011 Jul 15;27(14):1894-900. doi: 10.1093/bioinformatics/btr314. Epub 2011 May 26. Bioinformatics. 2011. PMID: 21622663
-
Exonization of transposed elements: A challenge and opportunity for evolution.Biochimie. 2011 Nov;93(11):1928-34. doi: 10.1016/j.biochi.2011.07.014. Epub 2011 Jul 26. Biochimie. 2011. PMID: 21787833 Review.
Cited by
-
ASPic-GeneID: a lightweight pipeline for gene prediction and alternative isoforms detection.Biomed Res Int. 2013;2013:502827. doi: 10.1155/2013/502827. Epub 2013 Nov 7. Biomed Res Int. 2013. PMID: 24308000 Free PMC article.
-
EGASP: the human ENCODE Genome Annotation Assessment Project.Genome Biol. 2006;7 Suppl 1(Suppl 1):S2.1-31. doi: 10.1186/gb-2006-7-s1-s2. Epub 2006 Aug 7. Genome Biol. 2006. PMID: 16925836 Free PMC article. Review.
-
Genome-wide analysis of differentially expressed genes and splicing isoforms in clear cell renal cell carcinoma.PLoS One. 2013 Oct 23;8(10):e78452. doi: 10.1371/journal.pone.0078452. eCollection 2013. PLoS One. 2013. PMID: 24194935 Free PMC article.
-
Identification of alternative 5'/3' splice sites based on the mechanism of splice site competition.Nucleic Acids Res. 2006;34(21):6305-13. doi: 10.1093/nar/gkl900. Epub 2006 Nov 10. Nucleic Acids Res. 2006. PMID: 17098928 Free PMC article.
-
ASPicDB: a database of annotated transcript and protein variants generated by alternative splicing.Nucleic Acids Res. 2011 Jan;39(Database issue):D80-5. doi: 10.1093/nar/gkq1073. Epub 2010 Nov 4. Nucleic Acids Res. 2011. PMID: 21051348 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
