

Integrated Modeling Program, Applied Chemical Theory (IMPACT)
- PMID: 16211539
- PMCID: PMC2742605
- DOI: 10.1002/jcc.20292
Integrated Modeling Program, Applied Chemical Theory (IMPACT)
Abstract
We provide an overview of the IMPACT molecular mechanics program with an emphasis on recent developments and a description of its current functionality. With respect to core molecular mechanics technologies we include a status report for the fixed charge and polarizable force fields that can be used with the program and illustrate how the force fields, when used together with new atom typing and parameter assignment modules, have greatly expanded the coverage of organic compounds and medicinally relevant ligands. As we discuss in this review, explicit solvent simulations have been used to guide our design of implicit solvent models based on the generalized Born framework and a novel nonpolar estimator that have recently been incorporated into the program. With IMPACT it is possible to use several different advanced conformational sampling algorithms based on combining features of molecular dynamics and Monte Carlo simulations. The program includes two specialized molecular mechanics modules: Glide, a high-throughput docking program, and QSite, a mixed quantum mechanics/molecular mechanics module. These modules employ the IMPACT infrastructure as a starting point for the construction of the protein model and assignment of molecular mechanics parameters, but have then been developed to meet specialized objectives with respect to sampling and the energy function.
(c) 2005 Wiley Periodicals, Inc.
Figures

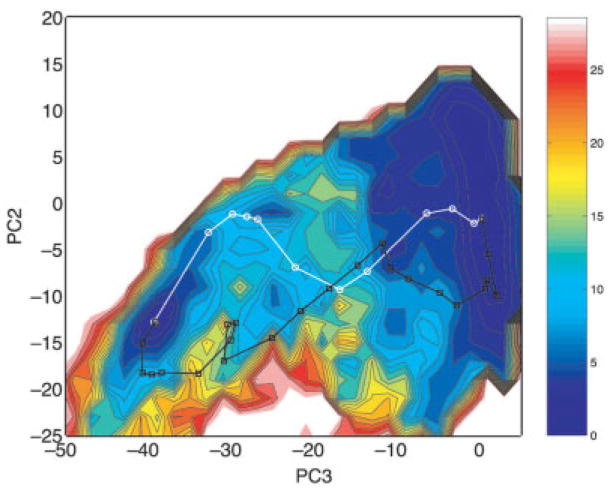
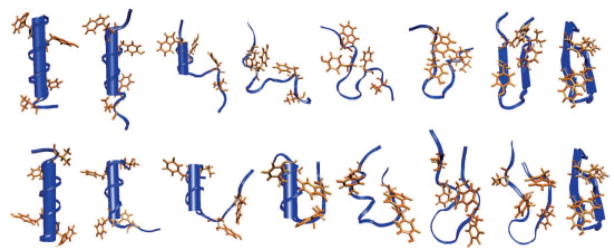
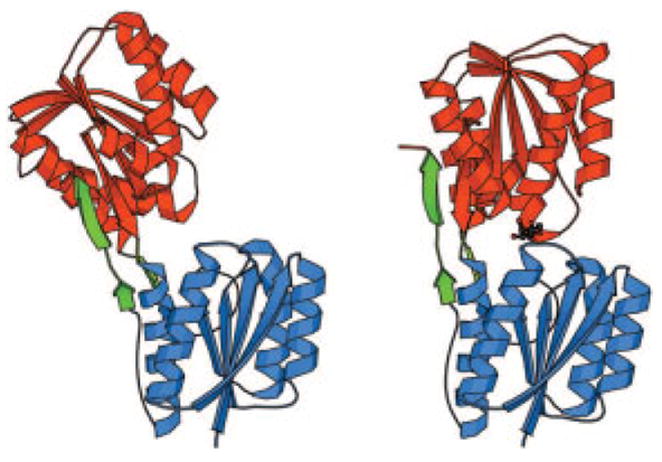
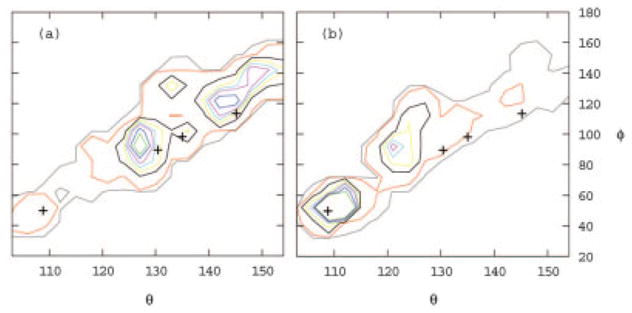
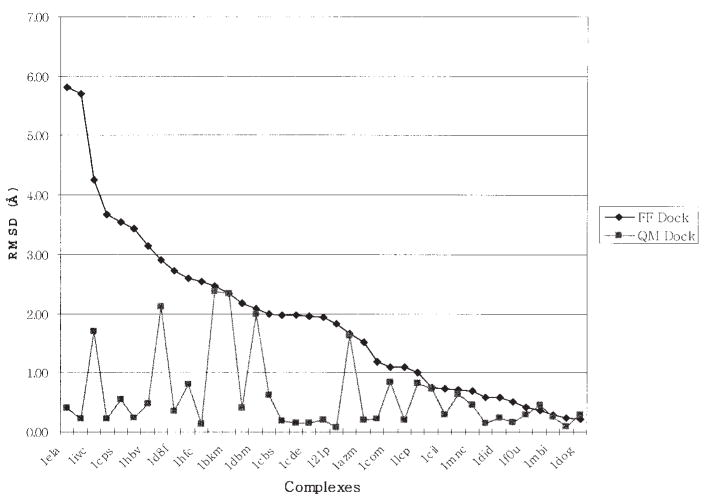
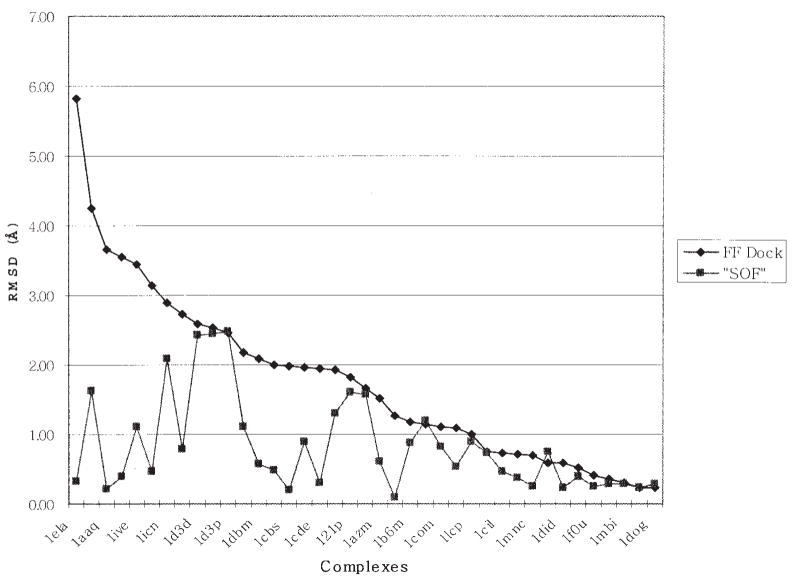
References
-
- McCammon JA, Gelin BR, Karplus M. Nature. 1977;267:585. - PubMed
-
- Karplus M. Biopolymers. 2003;68:350. - PubMed
-
- Levy RM, Bassolino D, Kitchen DB, Pardi A. Biochemistry. 1989;28:9361. - PubMed
-
- Fan P, Kominos D, Kitchen DB, Levy RM, Baum J. Chem Phys. 1991;158:295.
-
- Kitchen DB, Hirata F, Kofke DA, Westbrook JD, Yarmush M, Levy RM. J Comput Chem. 1990;11:1169.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
