

Protein molecular function prediction by Bayesian phylogenomics
- PMID: 16217548
- PMCID: PMC1246806
- DOI: 10.1371/journal.pcbi.0010045
Protein molecular function prediction by Bayesian phylogenomics
Abstract
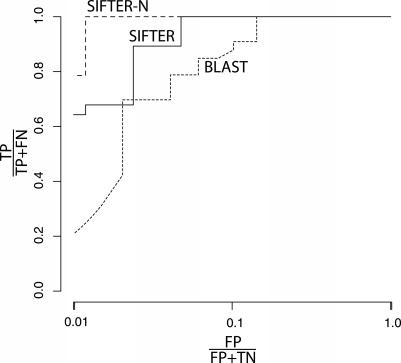
We present a statistical graphical model to infer specific molecular function for unannotated protein sequences using homology. Based on phylogenomic principles, SIFTER (Statistical Inference of Function Through Evolutionary Relationships) accurately predicts molecular function for members of a protein family given a reconciled phylogeny and available function annotations, even when the data are sparse or noisy. Our method produced specific and consistent molecular function predictions across 100 Pfam families in comparison to the Gene Ontology annotation database, BLAST, GOtcha, and Orthostrapper. We performed a more detailed exploration of functional predictions on the adenosine-5'-monophosphate/adenosine deaminase family and the lactate/malate dehydrogenase family, in the former case comparing the predictions against a gold standard set of published functional characterizations. Given function annotations for 3% of the proteins in the deaminase family, SIFTER achieves 96% accuracy in predicting molecular function for experimentally characterized proteins as reported in the literature. The accuracy of SIFTER on this dataset is a significant improvement over other currently available methods such as BLAST (75%), GeneQuiz (64%), GOtcha (89%), and Orthostrapper (11%). We also experimentally characterized the adenosine deaminase from Plasmodium falciparum, confirming SIFTER's prediction. The results illustrate the predictive power of exploiting a statistical model of function evolution in phylogenomic problems. A software implementation of SIFTER is available from the authors.
Conflict of interest statement
Figures





References
-
- Galperin MY, Koonin EV. Sources of systematic error in functional annotation of genomes: Domain rearrangement, non-orthologous gene displacement, and operon disruption. In Silico Biol. 1998;1:7. - PubMed
-
- Brenner SE. Errors in genome annotation. Trends Genet. 1999;15:132–133. - PubMed
-
- Koonin EV. Bridging the gap between sequence and function. Trends Genet. 2000;16:16. - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Khan S, Situ G, Decker K, Schmidt CJ. GoFigure: Automated gene ontology annotation. Bioinformatics. 2003;18:2484–2485. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
