

Scalable molecular dynamics with NAMD
- PMID: 16222654
- PMCID: PMC2486339
- DOI: 10.1002/jcc.20289
Scalable molecular dynamics with NAMD
Abstract
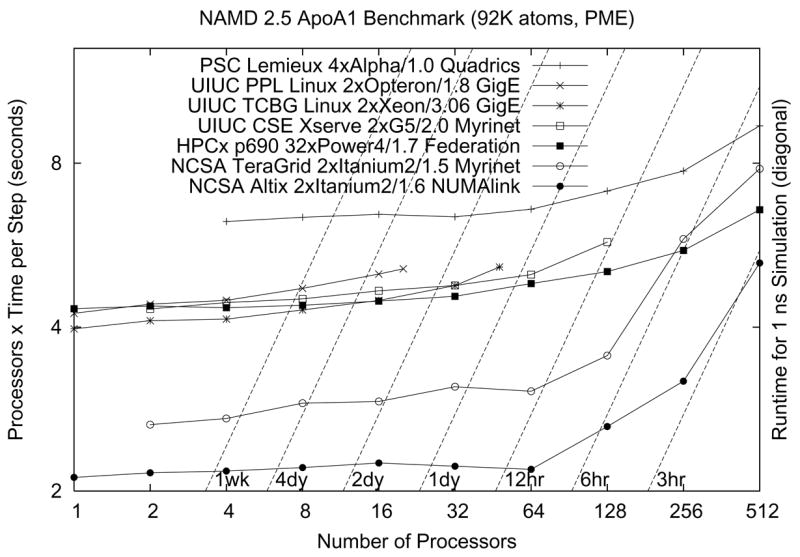
NAMD is a parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems. NAMD scales to hundreds of processors on high-end parallel platforms, as well as tens of processors on low-cost commodity clusters, and also runs on individual desktop and laptop computers. NAMD works with AMBER and CHARMM potential functions, parameters, and file formats. This article, directed to novices as well as experts, first introduces concepts and methods used in the NAMD program, describing the classical molecular dynamics force field, equations of motion, and integration methods along with the efficient electrostatics evaluation algorithms employed and temperature and pressure controls used. Features for steering the simulation across barriers and for calculating both alchemical and conformational free energy differences are presented. The motivations for and a roadmap to the internal design of NAMD, implemented in C++ and based on Charm++ parallel objects, are outlined. The factors affecting the serial and parallel performance of a simulation are discussed. Finally, typical NAMD use is illustrated with representative applications to a small, a medium, and a large biomolecular system, highlighting particular features of NAMD, for example, the Tcl scripting language. The article also provides a list of the key features of NAMD and discusses the benefits of combining NAMD with the molecular graphics/sequence analysis software VMD and the grid computing/collaboratory software BioCoRE. NAMD is distributed free of charge with source code at www.ks.uiuc.edu.
(c) 2005 Wiley Periodicals, Inc.
Figures

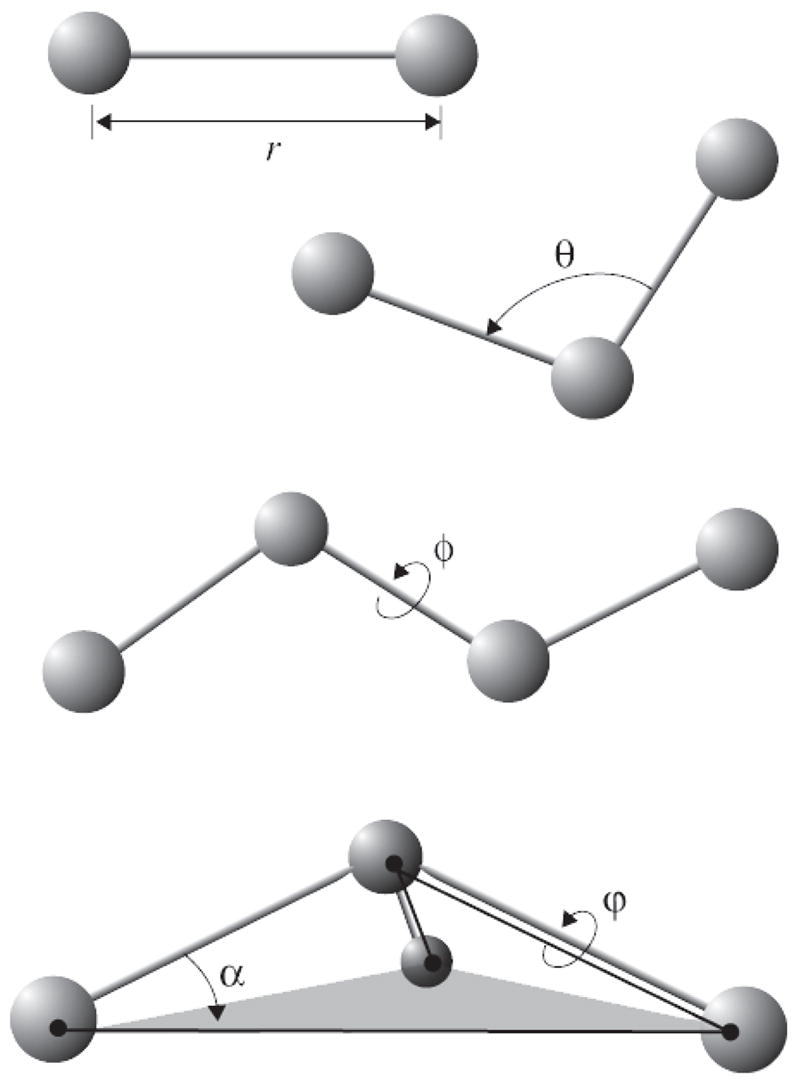
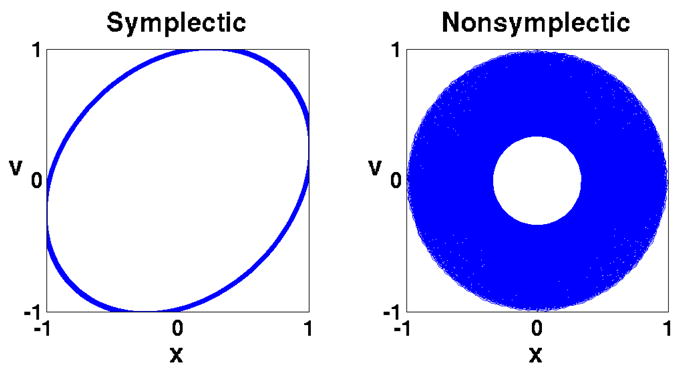


References
-
- Kalé Laxmikant, Skeel Robert, Bhandarkar Milind, Brunner Robert, Gursoy Attila, Krawetz Neal, Phillips James, Shinozaki Aritomo, Varadarajan Krishnan, Schulten Klaus. NAMD2: Greater scalability for parallel molecular dynamics. J Comp Phys. 1999;151:283–312.
-
- Humphrey William, Dalke Andrew, Schulten Klaus. VMD – Visual Molecular Dynamics. J Mol Graphics. 1996;14:33–38. - PubMed
-
- Nelson Mark, Humphrey William, Gursoy Attila, Dalke Andrew, Kalé Laxmikant, Skeel Robert, Schulten Klaus, Kufrin Richard. MDScope – A visual computing environment for structural biology. Comput Phys Commun. 1995;91(1–3):111–134.
-
- Nelson Mark, Humphrey William, Gursoy Attila, Dalke Andrew, Kalé Laxmikant, Skeel Robert D, Schulten Klaus. NAMD – A parallel, object-oriented molecular dynamics program. Int J Supercomp Appl High Perform Comp. 1996;10:251–268.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
