

Amino acid residues within conserved domain VI of the vesicular stomatitis virus large polymerase protein essential for mRNA cap methyltransferase activity
- PMID: 16227259
- PMCID: PMC1262600
- DOI: 10.1128/JVI.79.21.13373-13384.2005
Amino acid residues within conserved domain VI of the vesicular stomatitis virus large polymerase protein essential for mRNA cap methyltransferase activity
Abstract
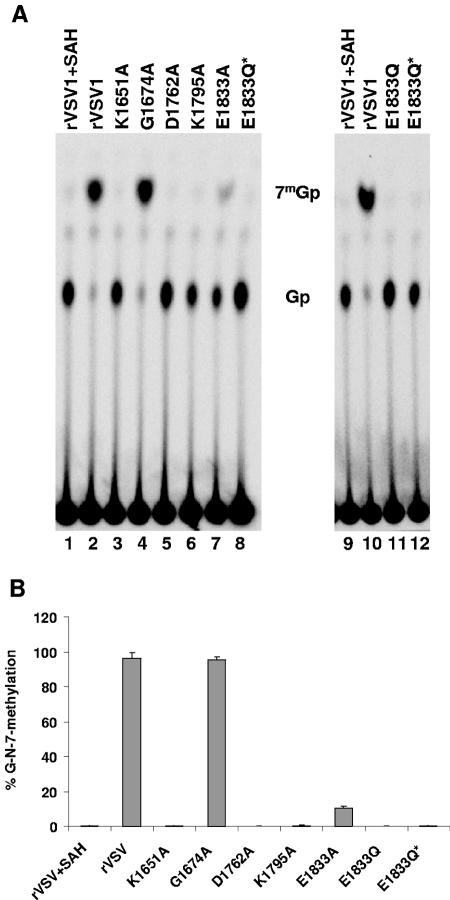
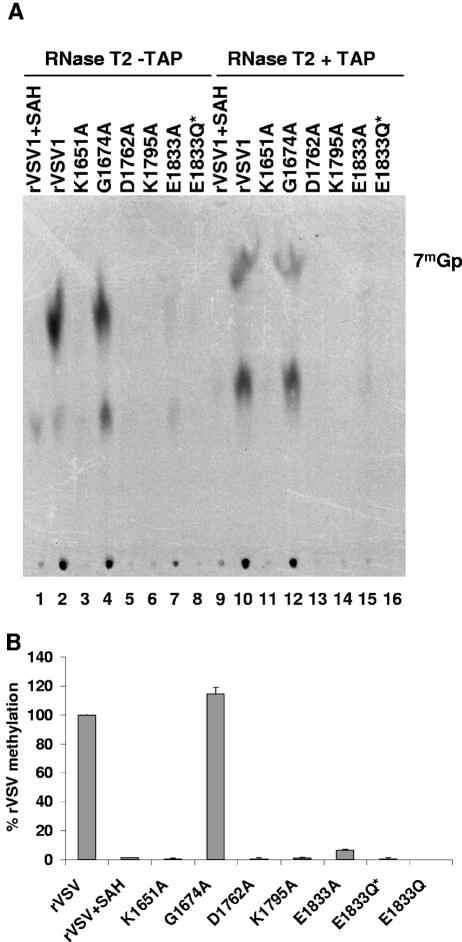
During mRNA synthesis, the polymerase of vesicular stomatitis virus (VSV) copies the genomic RNA to produce five capped and polyadenylated mRNAs with the 5'-terminal structure 7mGpppA(m)pApCpApGpNpNpApUpCp. The 5' mRNA processing events are poorly understood but presumably require triphosphatase, guanylyltransferase, [guanine-N-7]- and [ribose-2'-O]-methyltransferase (MTase) activities. Consistent with a role in mRNA methylation, conserved domain VI of the 241-kDa large (L) polymerase protein shares sequence homology with a bacterial [ribose-2'-O]-MTase, FtsJ/RrmJ. In this report, we generated six L gene mutations to test this homology. Individual substitutions to the predicted MTase active-site residues K1651, D1762, K1795, and E1833 yielded viruses with pinpoint plaque morphologies and 10- to 1,000-fold replication defects in single-step growth assays. Consistent with these defects, viral RNA and protein synthesis was diminished. In contrast, alteration of residue G1674 predicted to bind the methyl donor S-adenosylmethionine did not significantly perturb viral growth and gene expression. Analysis of the mRNA cap structure revealed that alterations to the predicted active site residues decreased [guanine-N-7]- and [ribose-2'-O]-MTase activity below the limit of detection of our assay. In contrast, the alanine substitution at G1674 had no apparent consequence. These data show that the predicted MTase active-site residues K1651, D1762, K1795, and E1833 within domain VI of the VSV L protein are essential for mRNA cap methylation. A model of mRNA processing consistent with these data is presented.
Figures
References
-
- Abraham, G., D. P. Rhodes, and A. K. Banerjee. 1975. The 5′ terminal structure of the methylated mRNA synthesized in vitro by vesicular stomatitis virus. Cell 5:51-58. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
