

Epsilon sarcoglycan mutations and phenotype in French patients with myoclonic syndromes
- PMID: 16227522
- PMCID: PMC2564513
- DOI: 10.1136/jmg.2005.036780
Epsilon sarcoglycan mutations and phenotype in French patients with myoclonic syndromes
Abstract
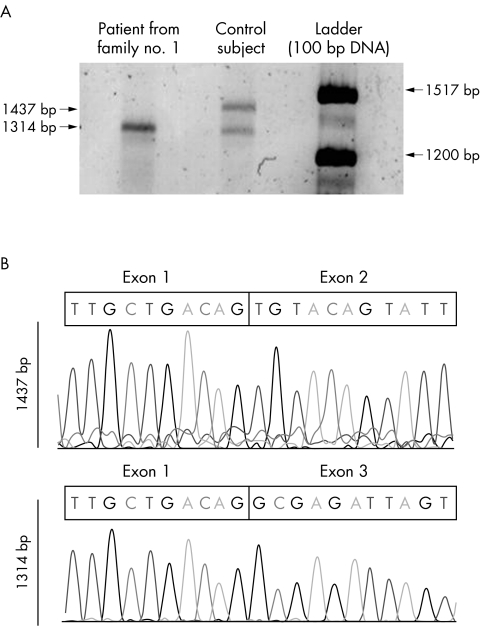
Background: Myoclonus dystonia syndrome (MDS) is an autosomal dominant movement disorder caused by mutations in the epsilon-sarcoglycan gene (SGCE) on chromosome 7q21.
Methods: We have screened for SGCE mutations in index cases from 76 French patients with myoclonic syndromes, including myoclonus dystonia (M-D), essential myoclonus (E-M), primary myoclonic dystonia, generalised dystonia, dystonia with tremor, and benign hereditary chorea. All coding exons of the SGCE gene were analysed. The DYT1 mutation was also tested.
Results: Sixteen index cases had SGCE mutations while one case with primary myoclonic dystonia carried the DYT1 mutation. Thirteen different mutations were found: three nonsense mutations, three missense mutations, three splice site mutations, three deletions, and one insertion. Eleven of the SGCE index cases had M-D and five E-M. No SGCE mutations were detected in patients with other phenotypes. The total number of mutation carriers in the families was 38, six of whom were asymptomatic. Penetrance was complete in paternal transmissions and null in maternal transmissions. MDS patients with SGCE mutation had a significantly earlier onset than the non-carriers. None of the patients had severe psychiatric disorders.
Conclusion: This large cohort of index patients shows that SGCE mutations are primarily found in patients with M-D and to a lesser extent E-M, but are present in only 30% of these patients combined (M-D and E-M).
Conflict of interest statement
Competing interests: none declared
References
-
- Asmus F, Gasser T. Inherited myoclonus‐dystonia. Adv Neurol 200494113–119. - PubMed
-
- Nygaard T G, Raymond D, Chen C, Nishino I, Greene P E, Jennings D, Heiman G A, Klein C, Saunders‐Pullman R J, Kramer P, Ozelius L J, Bressman S B. Localization of a gene for myoclonus‐dystonia to chromosome 7q21–q31. Ann Neurol 199946(5)794–798. - PubMed
-
- Zimprich A, Grabowski M, Asmus F, Naumann M, Berg D, Bertram M, Scheidtmann K, Kern P, Winkelmann J, Muller‐Myhsok B, Riedel L, Bauer M, Muller T, Castro M, Meitinger T, Strom T M, Gasser T. Mutations in the gene encoding epsilon‐sarcoglycan cause myoclonus‐dystonia syndrome. Nat Genet 200129(1)66–69. - PubMed
-
- Yoshida M, Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J Biochem (Tokyo) 1990108(5)748–752. - PubMed
-
- Bonnemann C G, Modi R, Noguchi S, Mizuno Y, Yoshida M, Gussoni E, McNally E M, Duggan D J, Angelini C, Hoffman E P. Beta‐sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet 199511(3)266–273. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous