

Natural history of Fabry disease in females in the Fabry Outcome Survey
- PMID: 16227523
- PMCID: PMC2563231
- DOI: 10.1136/jmg.2005.036327
Natural history of Fabry disease in females in the Fabry Outcome Survey
Abstract
Background: Fabry disease is a rare X linked lysosomal storage disorder resulting from deficiency of alpha-galactosidase A activity. Although the severity of clinical features in male patients is well described, only recently have studies reported the high prevalence of disabling clinical features in heterozygous females.
Aims: This study sets out to examine the clinical features and natural history of Fabry disease in further detail in a large group of female patients.
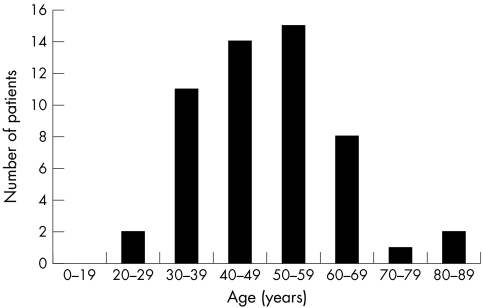
Methods: Data were obtained from 303 females enrolled in the Fabry Outcome Survey. Pain was assessed using the Brief Pain Inventory, and health related quality of life (HRQoL) was assessed using the European Quality of Life Questionnaire. A modified version of the Mainz Severity Score Index was also applied. Data on left ventricular mass (LVM) index, mean ventricular wall thickness, and glomerular filtration rate (GFR) were used to assess cardiac and renal involvement.
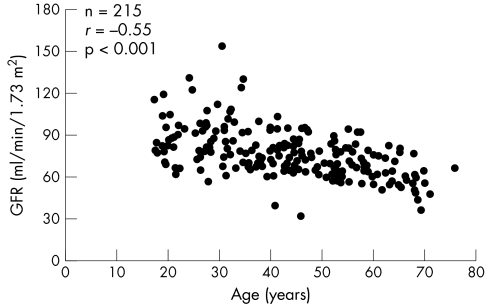
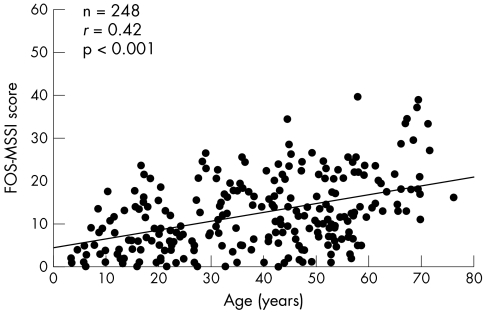
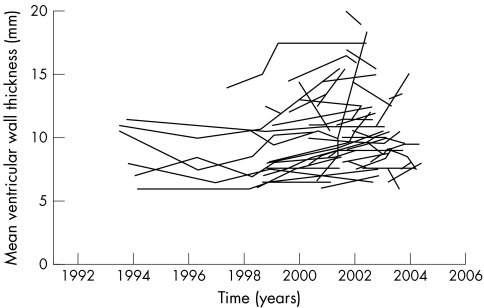
Results: The most commonly reported clinical features in females were neurological (77%) and cardiac (59%). A history of renal involvement was recorded in 40% of cases. Neurological features were the earliest to develop (mean age: 16 years), whereas cardiac (mean age: 33.5 years) and renal (mean age: 37.3 years) features developed later. LVM index increased exponentially with age. In addition, age was negatively correlated with estimated GFR and HRQoL.
Conclusions: Females with Fabry disease report important age related clinical features and clinical investigation demonstrates evidence of disease progression. This study highlights the importance of careful and longitudinal assessment of female heterozygote patients with Fabry disease.
Conflict of interest statement
Competing interests: P B Deegan, F Baehner, and D A Hughes have received honoraria and travel grants from TKT Europe. M‐Á Barba Romero has received honoraria and travel grants from TKT Europe and travel grants from Genzyme. M Beck has received honoraria, travel grants, and research grants from TKT Europe and Genzyme.

References
-
- Desnick R J, Ioannou Y A, Eng C M. α‐Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular basis of inherited disease. 8th ed. New York: McGraw‐Hill, 20013733–3774.
-
- Meikle P J, Hopwood J J, Clague A E, Carey W F. Prevalence of lysosomal storage disorders. JAMA 1999281249–254. - PubMed
-
- Kampmann C, Baehner F, Whybra C, Martin C, Wiethoff C M, Ries M, Gal A, Beck M. Cardiac manifestations of Anderson‐Fabry disease in heterozygous females. J Am Coll Cardiol 2002401668–1674. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical