

Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide
- PMID: 16227595
- PMCID: PMC1265832
- DOI: 10.1128/MCB.25.21.9447-9459.2005
Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide
Abstract
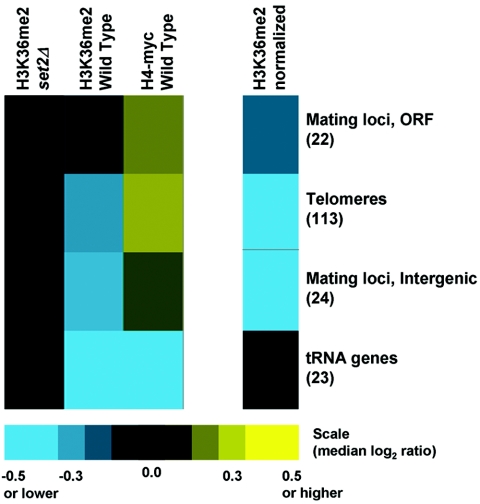
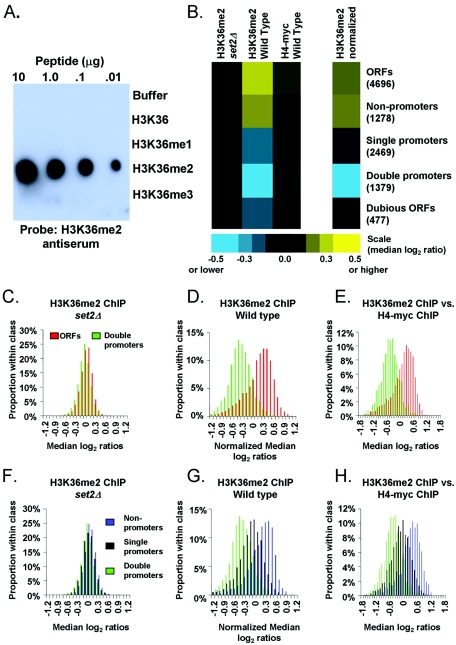
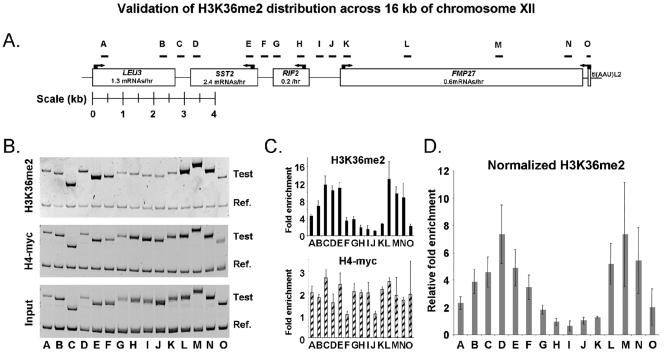
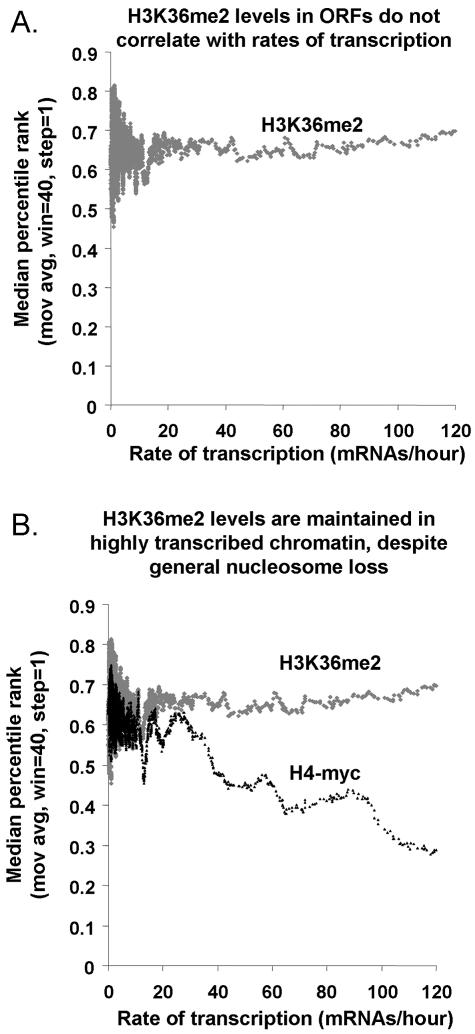
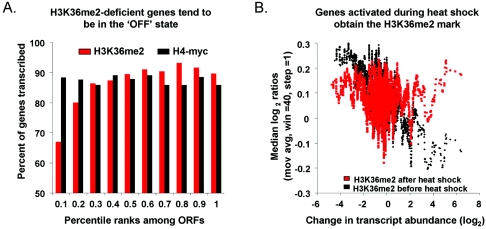
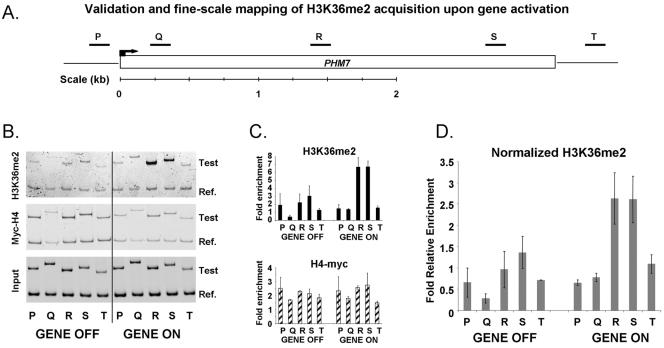
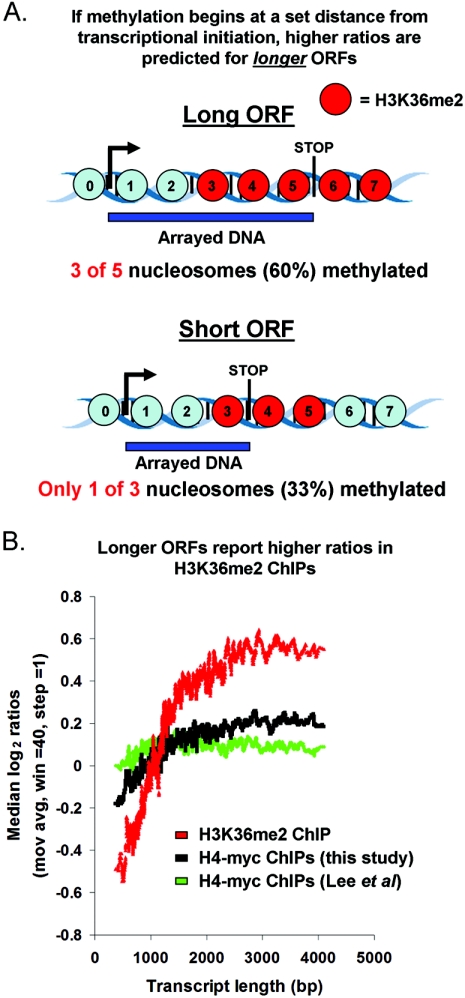
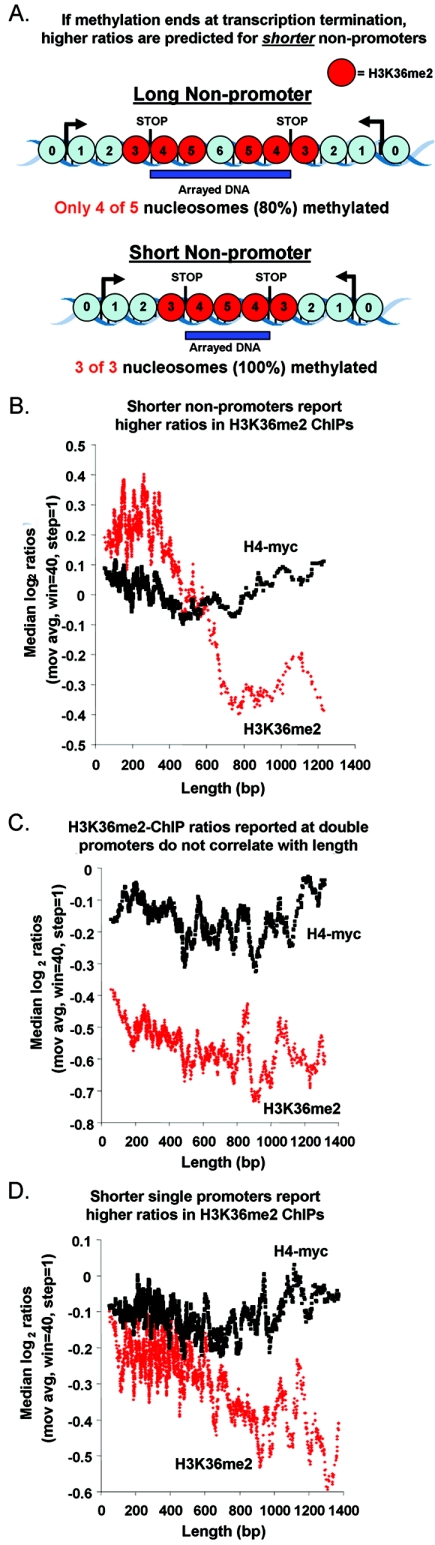
Set2p, which mediates histone H3 lysine 36 dimethylation (H3K36me2) in Saccharomyces cerevisiae, has been shown to associate with RNA polymerase II (RNAP II) at individual loci. Here, chromatin immunoprecipitation-microarray experiments normalized to general nucleosome occupancy reveal that nucleosomes within open reading frames (ORFs) and downstream noncoding chromatin were highly dimethylated at H3K36 and that Set2p activity begins at a stereotypic distance from the initiation of transcription genome-wide. H3K36me2 is scarce in regions upstream of divergently transcribed genes, telomeres, silenced mating loci, and regions transcribed by RNA polymerase III, providing evidence that the enzymatic activity of Set2p is restricted to its association with RNAP II. The presence of H3K36me2 within ORFs correlated with the "on" or "off" state of transcription, but the degree of H3K36 dimethylation within ORFs did not correlate with transcription frequency. This provides evidence that H3K36me2 is established during the initial instances of gene transcription, with subsequent transcription having at most a maintenance role. Accordingly, newly activated genes acquire H3K36me2 in a manner that does not correlate with gene transcript levels. Finally, nucleosomes dimethylated at H3K36 appear to be refractory to loss from highly transcribed chromatin. Thus, H3K36me2, which is highly conserved throughout eukaryotic evolution, provides a stable molecular mechanism for establishing chromatin context throughout the genome by distinguishing potential regulatory regions from transcribed chromatin.
Figures
References
-
- Bannister, A. J., R. Schneider, F. A. Myers, A. W. Thorne, C. Crane-Robinson, and T. Kouzarides. 2005. Spatial distribution of di-and tri-methyl lysine 36 of histone H3 at active genes. J. Biol. Chem. 280:17732-17736. - PubMed
-
- Boeger, H., J. Griesenbeck, J. S. Strattan, and R. D. Kornberg. 2003. Nucleosomes unfold completely at a transcriptionally active promoter. Mol. Cell 11:1587-1598. - PubMed
-
- Boeger, H., J. Griesenbeck, J. S. Strattan, and R. D. Kornberg. 2004. Removal of promoter nucleosomes by disassembly rather than sliding in vivo. Mol. Cell 14:667-673. - PubMed
-
- Bohlander, S. K., R. Espinosa III, M. M. Le Beau, J. D. Rowley, and M. O. Diaz. 1992. A method for the rapid sequence-independent amplification of microdissected chromosomal material. Genomics 13:1322-1324. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases