

Full motor recovery despite striatal neuron loss and formation of irreversible amyloid-like inclusions in a conditional mouse model of Huntington's disease
- PMID: 16237181
- PMCID: PMC6725739
- DOI: 10.1523/JNEUROSCI.3183-05.2005
Full motor recovery despite striatal neuron loss and formation of irreversible amyloid-like inclusions in a conditional mouse model of Huntington's disease
Abstract
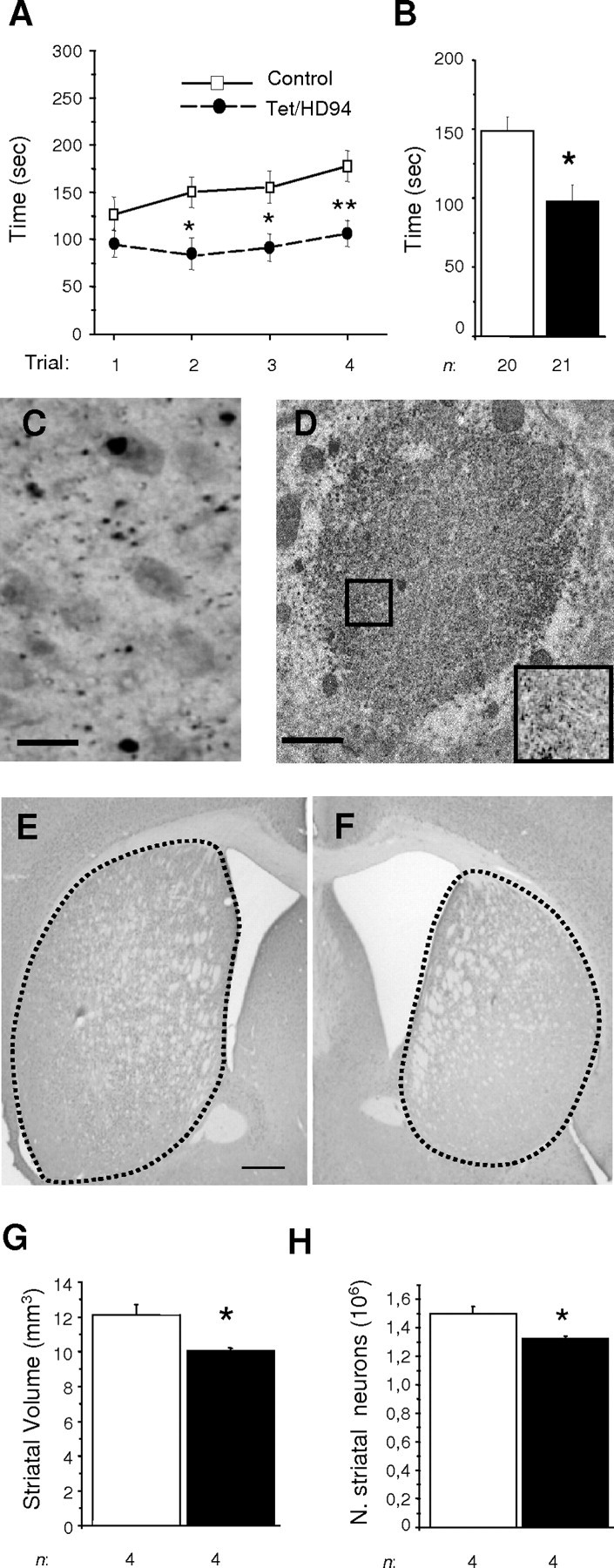
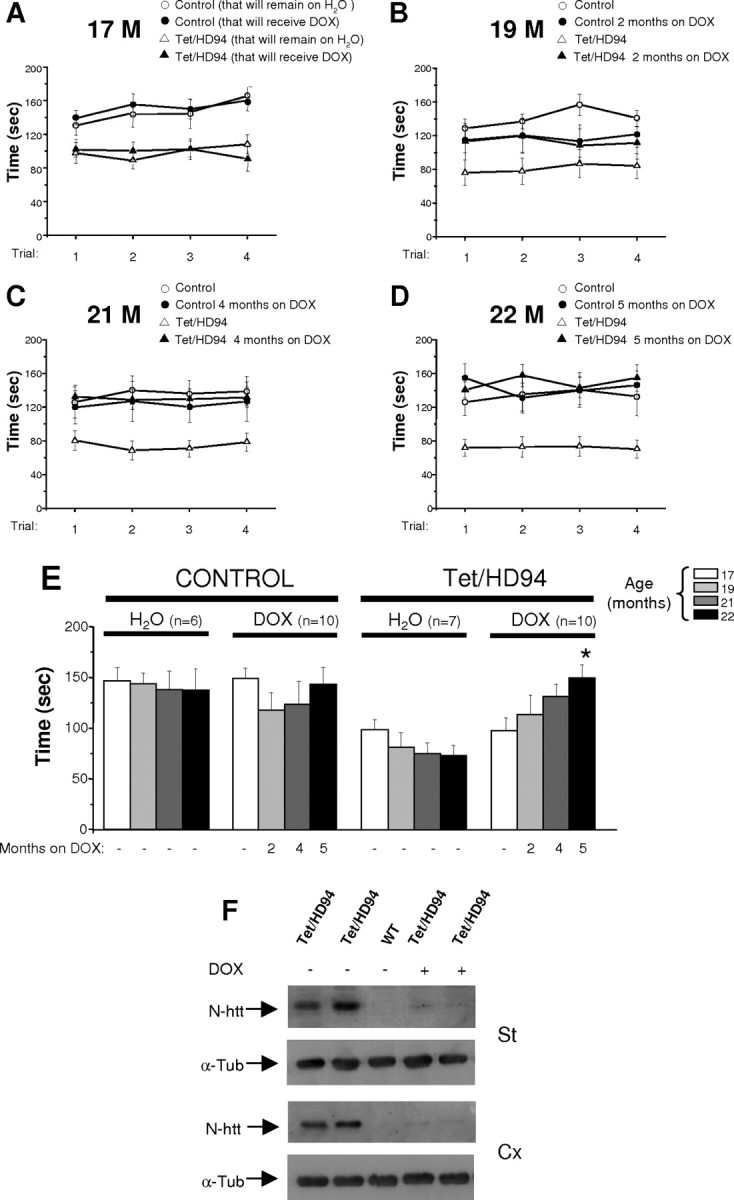
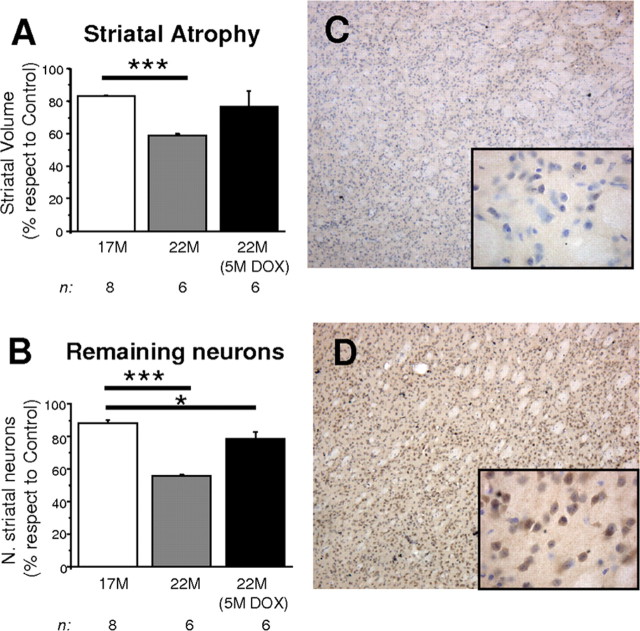
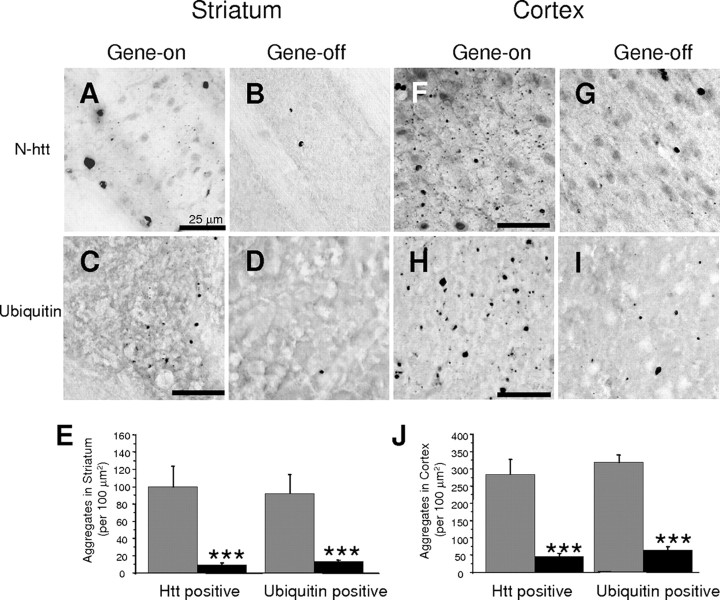
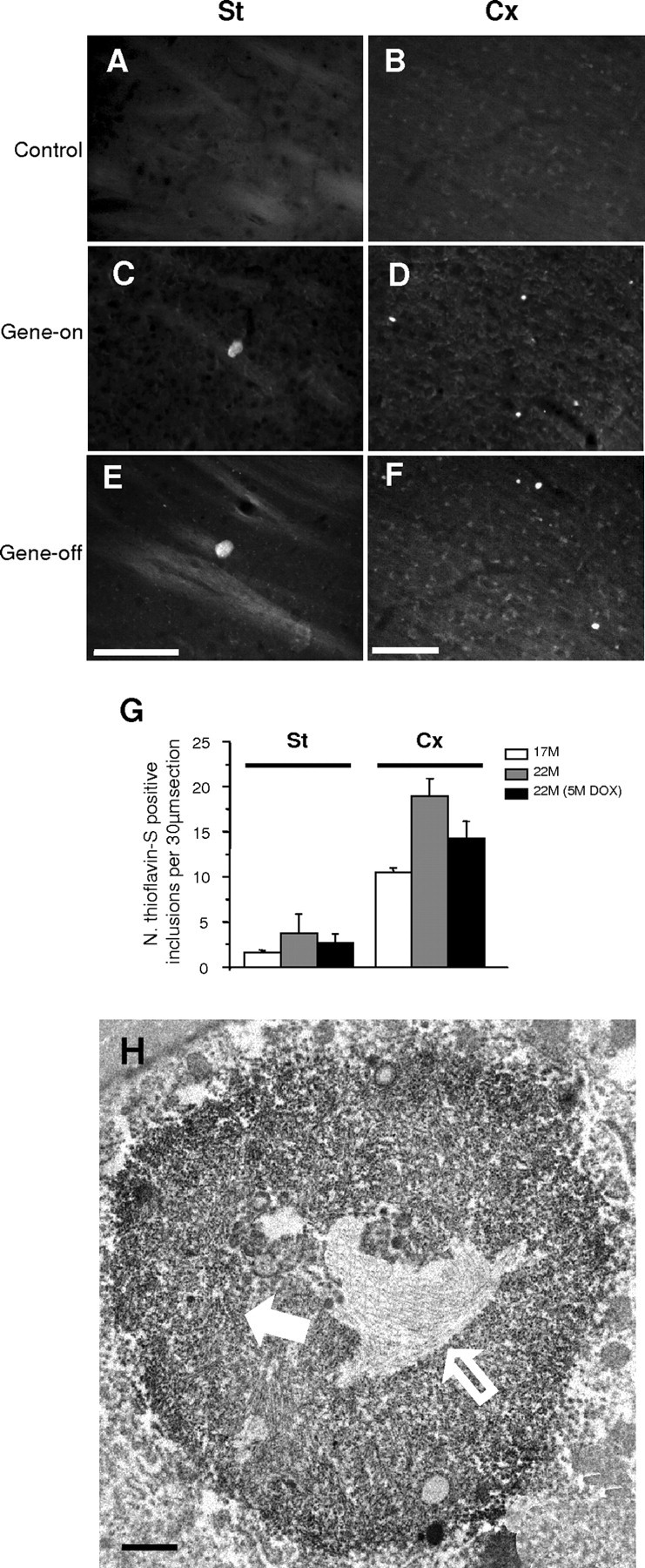
The primary mechanism responsible for Huntington's disease remains unknown. Postulated early pathogenic events include the following: impaired protein folding, altered protein degradation, mitochondrial dysfunction, and transcriptional dysregulation. Although related therapies can delay disease progression in mouse models, they target downstream and probably indirect effects of mutant-huntingtin expression. Accordingly, in case they prove beneficial in humans, they might only palliate some aspects of disease. Our previous studies in the Tet/HD94 conditional model and the recently reported efficacy of RNA interference against mutant huntingtin in another mouse model support silencing mutant-huntingtin expression as a valid therapeutic approach that has the advantage of targeting toxicity at its root. Here, we address whether gene silencing can still be beneficial in the late stages of disease with detectable striatal neuron loss. Stereological analysis was applied to determine an age at which Tet/HD94 mice show a decrease in the number of striatal neurons. Then, progression of neuropathology and motor phenotype were analyzed in mice that were allowed to continue expressing mutant huntingtin and in mice that no longer expressed it. Neuronal loss did not revert in gene-off mice, but the additional loss that takes place in gene-on mice was prevented. The total number of huntingtin-containing inclusions dramatically reverted, but a small fraction of inclusions positive for the amyloid dye thioflavin-S remained. Interestingly, despite a 20% decrease in striatal neurons and the presence of amyloid-like irreversible inclusions, gene-off mice fully recover from their motor deficit, thus ruling out amyloid-like huntingtin inclusions as the main toxic species and suggesting that gene-silencing therapies might work in late stages of disease.
Figures
References
-
- Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, Friedlich A, Browne SE, Schilling G, Borchelt DR, Hersch SM, Ross CA, Beal MF (2001) Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol Dis 8: 479-491. - PubMed
-
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431: 805-810. - PubMed
-
- Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martin-Ibanez R, Munoz MT, Mengod G, Ernfors P, Alberch J (2004) Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington's disease. J Neurosci 24: 7727-7739. - PMC - PubMed
-
- Chen J, Kelz MB, Zeng G, Sakai N, Steffen C, Shockett PE, Picciotto MR, Duman RS, Nestler EJ (1998) Transgenic animals with inducible, targeted gene expression in brain. Mol Pharmacol 54: 495-503. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases