

Mitochondrial criticality: a new concept at the turning point of life or death
- PMID: 16242921
- PMCID: PMC2692535
- DOI: 10.1016/j.bbadis.2005.06.008
Mitochondrial criticality: a new concept at the turning point of life or death
Abstract
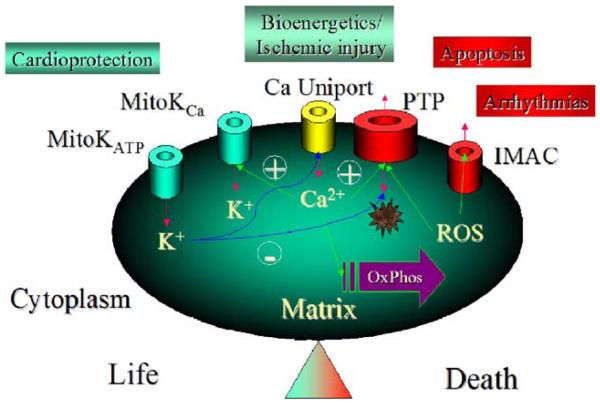
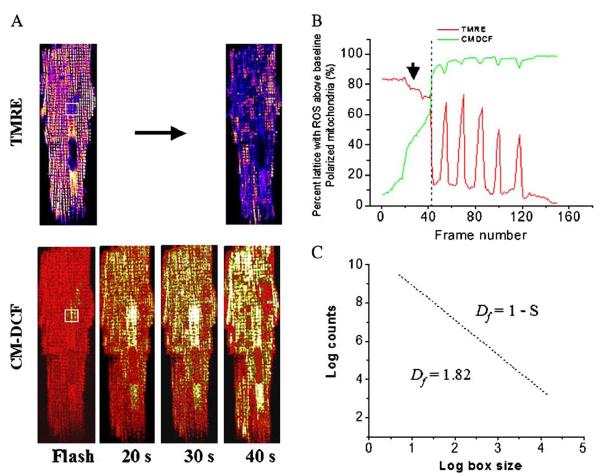
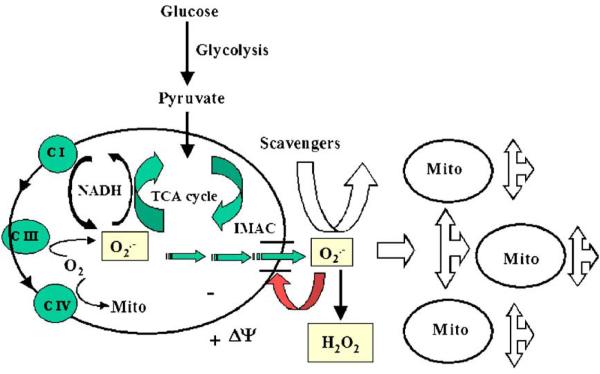
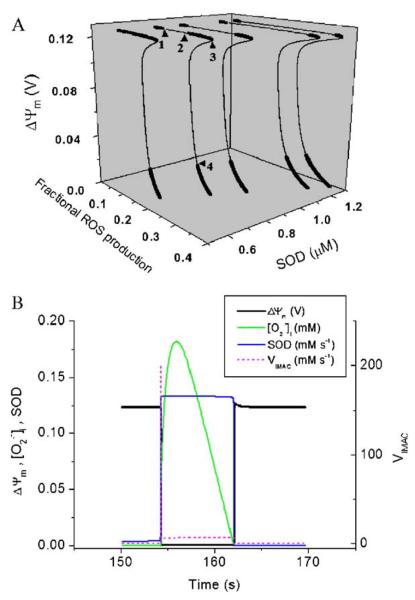
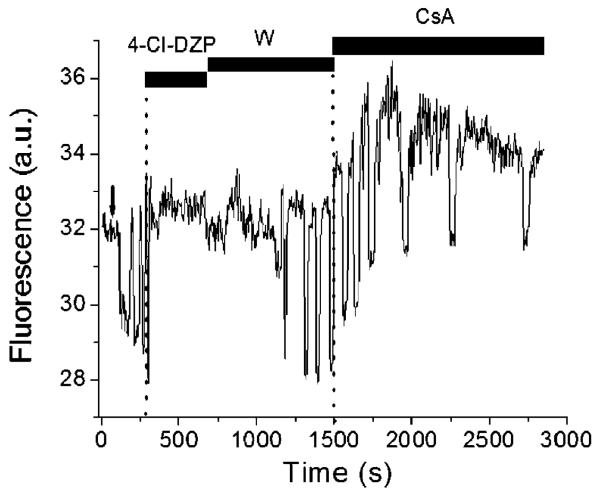
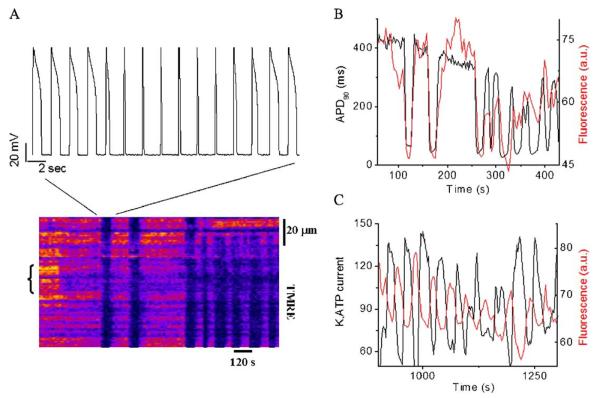
A variety of stressors can cause the collapse of mitochondrial membrane potential (DeltaPsi(m)), but the events leading up to this catastrophic cellular event are not well understood at the mechanistic level. Based on our recent studies of oscillations in mitochondrial energetics, we have coined the term "mitochondrial criticality" to describe the state in which the mitochondrial network of cardiomyocytes becomes very sensitive to small perturbations in reactive oxygen species (ROS), resulting in the scaling of local mitochondrial uncoupling and DeltaPsi(m) loss to the whole cell, and the myocardial syncytium. At the point of criticality, the dynamics of the mitochondrial network bifurcate to oscillatory behavior. These energetic changes are translated into effects on the electrical excitability of the cell, inducing dramatic changes in the morphology and the threshold for activating an action potential. Emerging evidence suggests that this mechanism, by creating spatial and temporal heterogeneity of excitability in the heart during ischemia and reperfusion, underlies the genesis of potentially lethal cardiac arrhythmias.
Figures
References
-
- Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. - PubMed
-
- Nicholls DG, Ferguson SJ. Bioenergetics. 3rd ed. Vol. 3. Academic Press; 2002.
-
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 1990;258:C755–C786. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
