

Protein-DNA binding specificity predictions with structural models
- PMID: 16246914
- PMCID: PMC1270944
- DOI: 10.1093/nar/gki875
Protein-DNA binding specificity predictions with structural models
Abstract
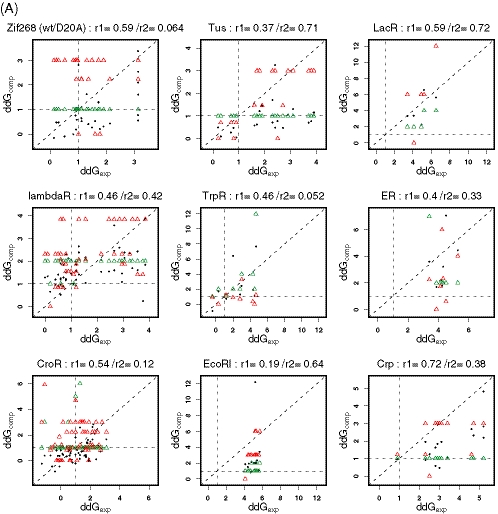
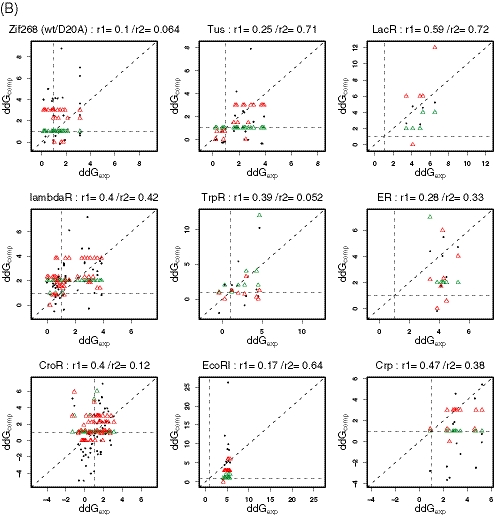
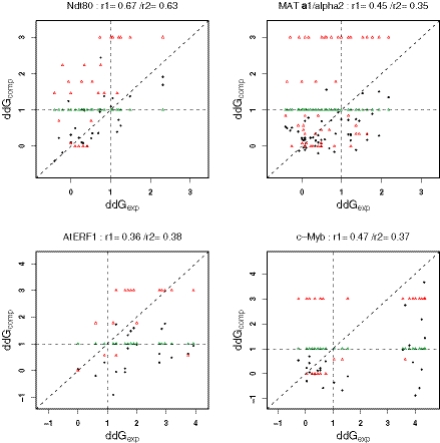
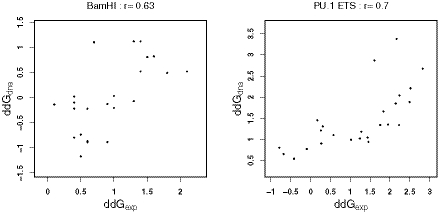
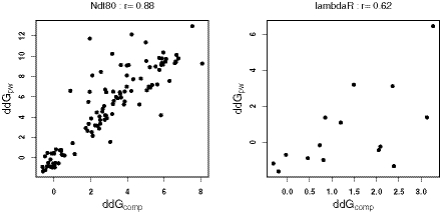
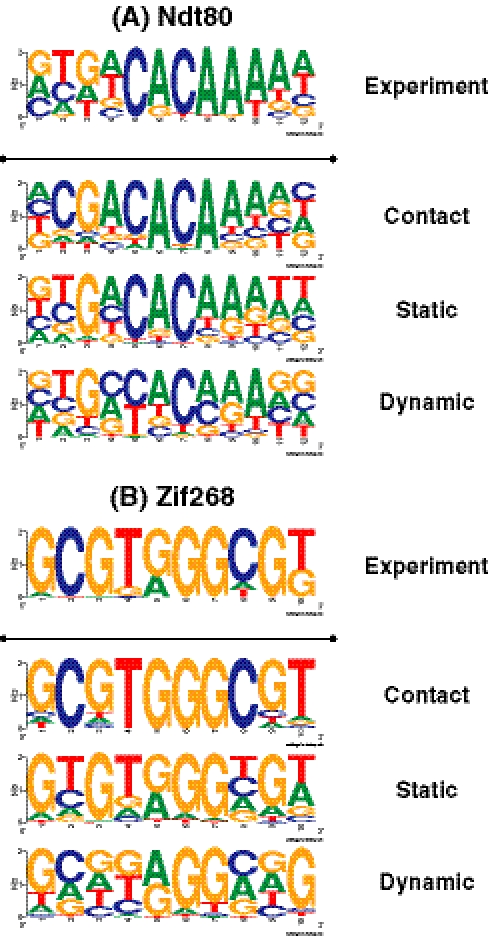
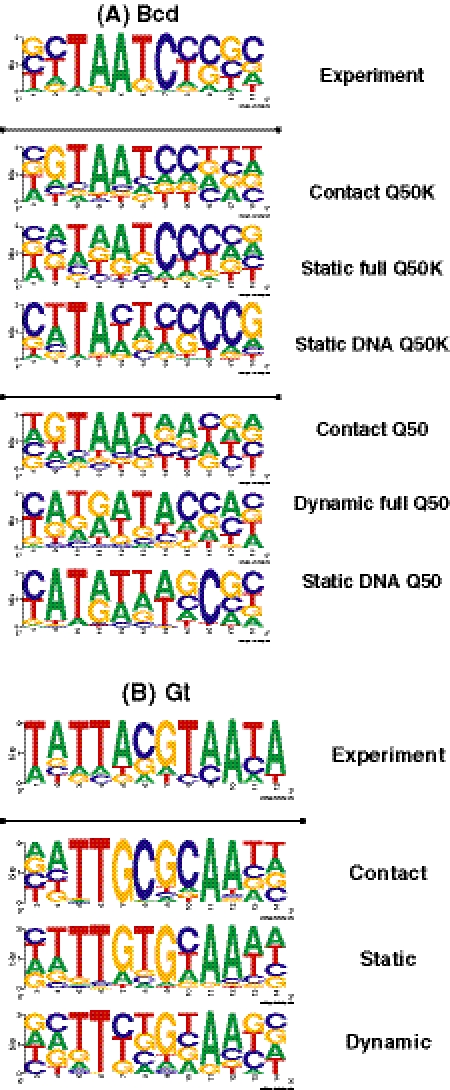
Protein-DNA interactions play a central role in transcriptional regulation and other biological processes. Investigating the mechanism of binding affinity and specificity in protein-DNA complexes is thus an important goal. Here we develop a simple physical energy function, which uses electrostatics, solvation, hydrogen bonds and atom-packing terms to model direct readout and sequence-specific DNA conformational energy to model indirect readout of DNA sequence by the bound protein. The predictive capability of the model is tested against another model based only on the knowledge of the consensus sequence and the number of contacts between amino acids and DNA bases. Both models are used to carry out predictions of protein-DNA binding affinities which are then compared with experimental measurements. The nearly additive nature of protein-DNA interaction energies in our model allows us to construct position-specific weight matrices by computing base pair probabilities independently for each position in the binding site. Our approach is less data intensive than knowledge-based models of protein-DNA interactions, and is not limited to any specific family of transcription factors. However, native structures of protein-DNA complexes or their close homologs are required as input to the model. Use of homology modeling can significantly increase the extent of our approach, making it a useful tool for studying regulatory pathways in many organisms and cell types.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
