

Building native protein conformation from highly approximate backbone torsion angles
- PMID: 16251268
- PMCID: PMC1283474
- DOI: 10.1073/pnas.0508415102
Building native protein conformation from highly approximate backbone torsion angles
Abstract
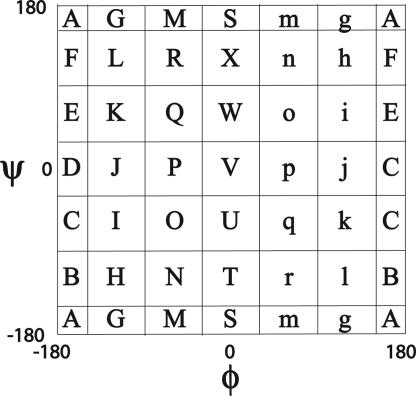
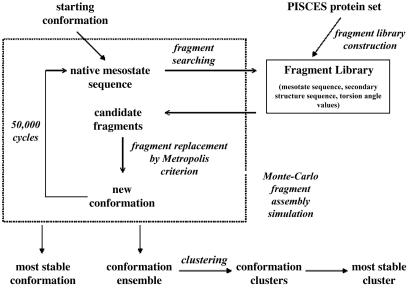
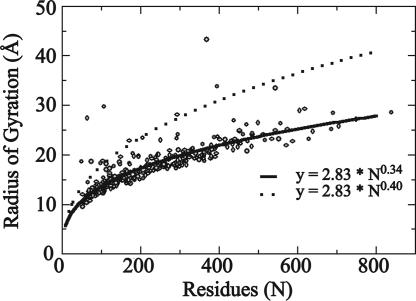
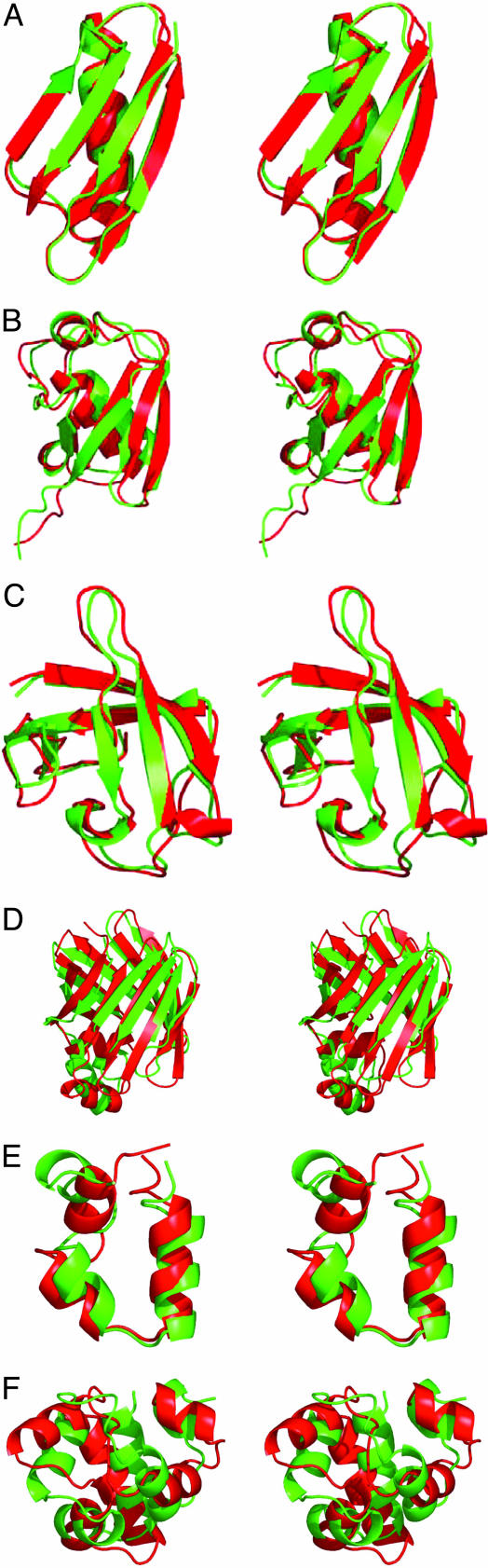
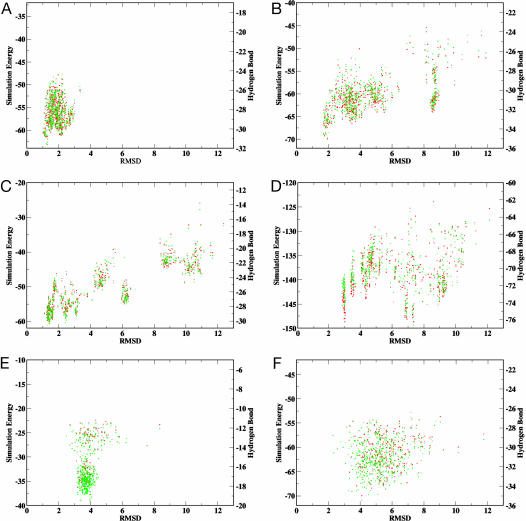
Reconstructing a protein in three dimensions from its backbone torsion angles is an ongoing challenge because minor inaccuracies in these angles produce major errors in the structure. As a familiar example, a small change in an elbow angle causes a large displacement at the end of your arm, the longer the arm, the larger the displacement. Even accurate knowledge of the backbone torsions and Psi is insufficient, owing to the small, but cumulative, deviations from ideality in backbone planarity, which, if ignored, also lead to major errors in the structure. Against this background, we conducted a computational experiment to assess whether protein conformation can be determined from highly approximate backbone torsion angles, the kind of information that is now obtained readily from NMR. Specifically, backbone torsion angles were taken from proteins of known structure and mapped into 60 degrees x 60 degrees grid squares, called mesostates. Side-chain atoms beyond the beta -carbon were discarded. A mesostate representation of the protein backbone was then used to extract likely candidates from a fragment library of mesostate pentamers, followed by Monte Carlo-based fragment-assembly simulations to identify stable conformations compatible with the given mesostate sequence. Only three simple energy terms were used to gauge stability: molecular compaction, soft-sphere repulsion, and hydrogen bonding. For the six representative proteins described here, stable conformers can be partitioned into a remarkably small number of topologically distinct clusters. Among these, the native topology is found with high frequency and can be identified as the cluster with the most favorable energy.
Figures
Similar articles
-
Secondary structure determines protein topology.Protein Sci. 2006 Aug;15(8):1829-34. doi: 10.1110/ps.062305106. Epub 2006 Jul 5. Protein Sci. 2006. PMID: 16823044 Free PMC article.
-
Building native protein conformation from NMR backbone chemical shifts using Monte Carlo fragment assembly.Protein Sci. 2007 Aug;16(8):1515-21. doi: 10.1110/ps.072988407. Protein Sci. 2007. PMID: 17656574 Free PMC article.
-
A Monte Carlo method for generating structures of short single-stranded DNA sequences.Biopolymers. 1993 Jan;33(1):75-105. doi: 10.1002/bip.360330109. Biopolymers. 1993. PMID: 8427940
-
Dynamic Monte Carlo simulations of a new lattice model of globular protein folding, structure and dynamics.J Mol Biol. 1991 Sep 20;221(2):499-531. doi: 10.1016/0022-2836(91)80070-b. J Mol Biol. 1991. PMID: 1920430 Review.
-
Intramolecular backbone···backbone hydrogen bonds in polypeptide conformations. The other way around: ɛ-turn.Biopolymers. 2017 Jan;108(1). doi: 10.1002/bip.22911. Biopolymers. 2017. PMID: 27404945 Review.
Cited by
-
Fragment-HMM: a new approach to protein structure prediction.Protein Sci. 2008 Nov;17(11):1925-34. doi: 10.1110/ps.036442.108. Epub 2008 Aug 22. Protein Sci. 2008. PMID: 18723665 Free PMC article.
-
Electrostatic solvation energy for two oppositely charged ions in a solvated protein system: salt bridges can stabilize proteins.Biophys J. 2010 Feb 3;98(3):470-7. doi: 10.1016/j.bpj.2009.10.031. Biophys J. 2010. PMID: 20141761 Free PMC article.
-
Assessing the solvent-dependent surface area of unfolded proteins using an ensemble model.Proc Natl Acad Sci U S A. 2008 Mar 4;105(9):3321-6. doi: 10.1073/pnas.0712240105. Epub 2008 Feb 27. Proc Natl Acad Sci U S A. 2008. PMID: 18305164 Free PMC article.
-
Influence of nonlinear electrostatics on transfer energies between liquid phases: charge burial is far less expensive than Born model.Proc Natl Acad Sci U S A. 2008 Aug 12;105(32):11146-51. doi: 10.1073/pnas.0804506105. Epub 2008 Aug 4. Proc Natl Acad Sci U S A. 2008. PMID: 18678891 Free PMC article.
-
Predicting continuous local structure and the effect of its substitution for secondary structure in fragment-free protein structure prediction.Structure. 2009 Nov 11;17(11):1515-27. doi: 10.1016/j.str.2009.09.006. Structure. 2009. PMID: 19913486 Free PMC article.
References
-
- Anfinsen, C. B. (1973) Science 181, 223-230. - PubMed
-
- Fitzkee, N. C. & Rose, G. D. (2005) J. Mol. Biol., in press. - PubMed
-
- DePristo, M. A., de Bakker, P. I. & Blundell, T. L. (2004) Structure (London) 12, 831-838. - PubMed
-
- Fitzkee, N. C., Fleming, P. J., Gong, H., Panasik, N., Jr., Street, T. O. & Rose, G. D. (2005) Trends Biochem. Sci. 30, 73-80. - PubMed
-
- Shortle, D. (2002) Adv. Protein Chem. 62, 1-23. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
