

Na+-dependent sources of intra-axonal Ca2+ release in rat optic nerve during in vitro chemical ischemia
- PMID: 16251444
- PMCID: PMC6725557
- DOI: 10.1523/JNEUROSCI.2003-05.2005
Na+-dependent sources of intra-axonal Ca2+ release in rat optic nerve during in vitro chemical ischemia
Abstract
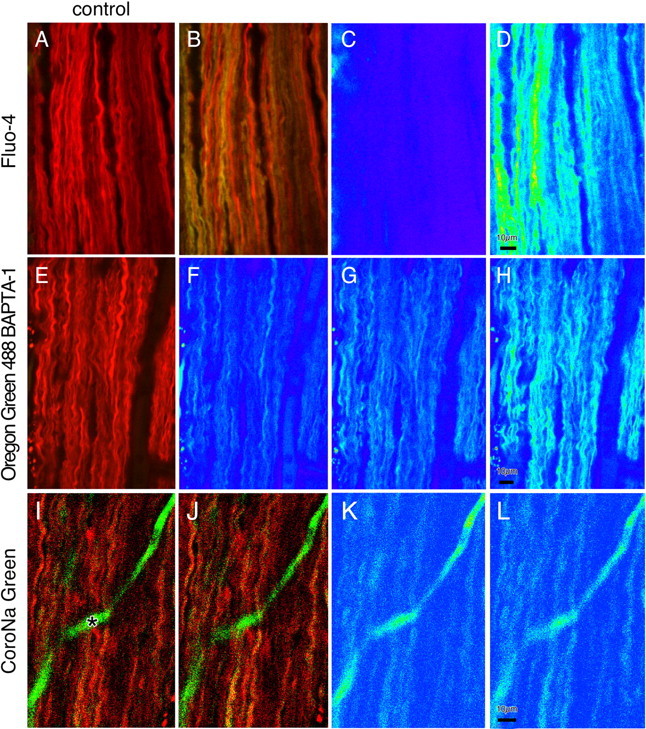
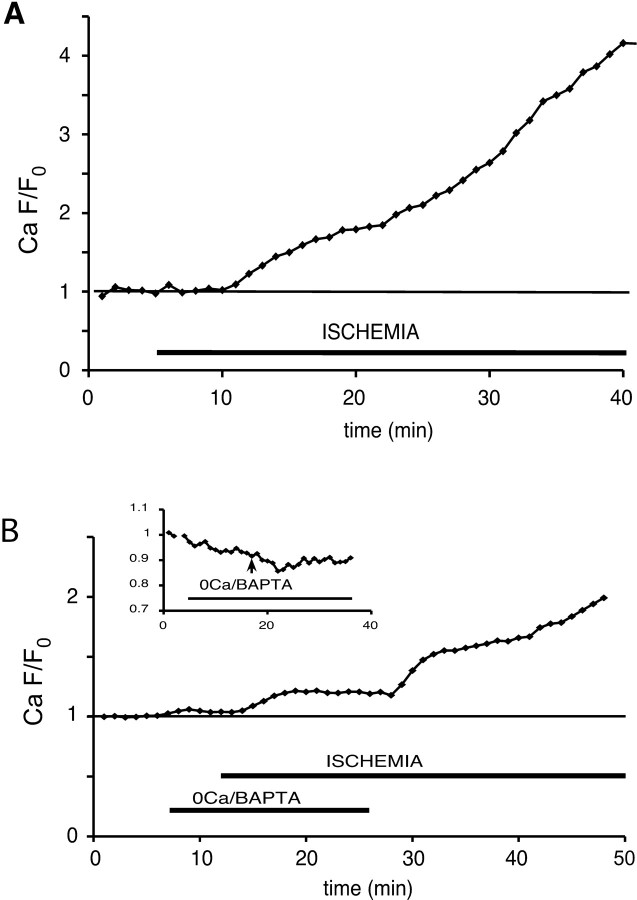
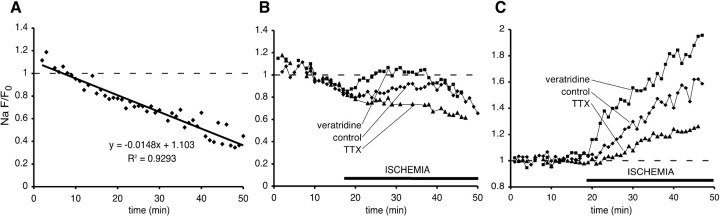
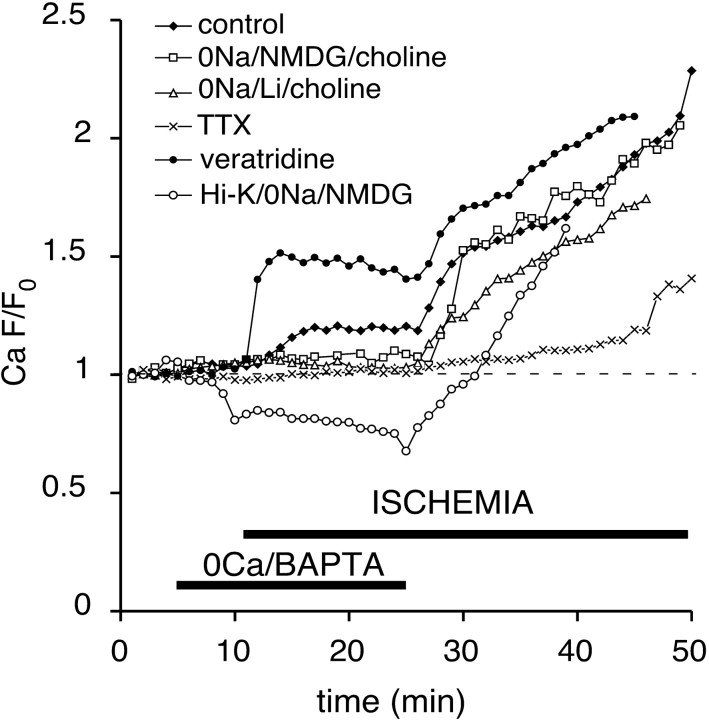
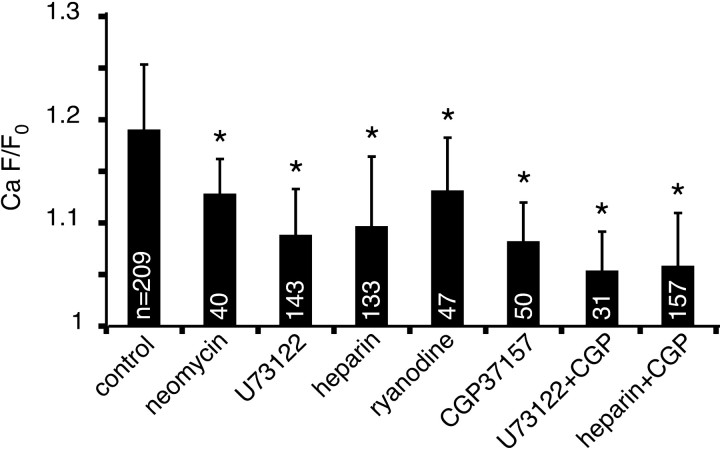
The contribution of intracellular stores to axonal Ca2+ overload during chemical ischemia in vitro was examined by confocal microscopy. Ca2+ accumulation was measured by fluo-4 dextran (low-affinity dye, KD approximately 4 microM) or by Oregon Green 488 BAPTA-1 dextran (highaffinity dye, KD approximately 450 nM). Axonal Na+ was measured using CoroNa Green. Ischemia in CSF containing 2 mM Ca2+ caused an approximately 3.5-fold increase in fluo-4 emission after 30 min, indicating a large axonal Ca2+ rise well into the micromolar range. Axonal Na+ accumulation was enhanced by veratridine and reduced, but not abolished, by TTX. Ischemia in Ca2+-free (plus BAPTA) perfusate resulted in a smaller but consistent Ca2+ increase monitored by Oregon Green 488 BAPTA-1, indicating release from intracellular sources. This release was eliminated in large part when Na+ influx was reduced by replacement with N-methyl-D-glucamine (NMDG+; even in depolarizing high K+ perfusate), Li+, or by the application of TTX and significantly increased by veratridine. Intracellular release also was reduced significantly by neomycin or 1-(6-[(17beta-methoxyestra-1,3,5 [10]-trien-17-yl) amino] hexyl)-1H-pyrrole-2,5-dione (U73122 [GenBank]) (phospholipase C inhibitors), heparin [inositol trisphosphate (IP3) receptor blocker], or 7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP37157; mitochondrial Na+/Ca2+ exchange inhibitor) as well as ryanodine. Combining CGP37157 with U73122 [GenBank] or heparin decreased the response more than either agent alone and significantly improved electrophysiological recovery. Our conclusion is that intra-axonal Ca2+ release during ischemia in rat optic nerve is mainly dependent on Na+ influx. This Na+ accumulation stimulates three distinct intra-axonal sources of Ca2+: (1) the mitochondrial Na+/Ca2+ exchanger driven in the Na+ import/Ca2+ export mode, (2) positive modulation of ryanodine receptors, and (3) promotion of IP3 generation by phospholipase C.
Figures
Similar articles
-
Sources of axonal calcium loading during in vitro ischemia of rat dorsal roots.Muscle Nerve. 2007 Apr;35(4):451-7. doi: 10.1002/mus.20731. Muscle Nerve. 2007. PMID: 17206661
-
Coupling of calcium homeostasis to axonal sodium in axons of mouse optic nerve.J Neurophysiol. 2002 Aug;88(2):802-16. doi: 10.1152/jn.2002.88.2.802. J Neurophysiol. 2002. PMID: 12163532
-
Release of Ca2+ is the crucial step for the potentiation of IPSCs in the cultured cerebellar Purkinje cells of the rat.J Physiol. 1996 Dec 15;497 ( Pt 3)(Pt 3):611-27. doi: 10.1113/jphysiol.1996.sp021794. J Physiol. 1996. PMID: 9003548 Free PMC article.
-
Excessive release of [3H] noradrenaline by veratridine and ischemia in spinal cord.Neurochem Int. 2001 Jul;39(1):59-63. doi: 10.1016/s0197-0186(00)00124-8. Neurochem Int. 2001. PMID: 11311450
-
Fundamental importance of Na+-Ca2+ exchange for the pacemaking mechanism in guinea-pig sino-atrial node.J Physiol. 2006 Mar 15;571(Pt 3):639-49. doi: 10.1113/jphysiol.2005.100305. Epub 2006 Jan 19. J Physiol. 2006. PMID: 16423859 Free PMC article.
Cited by
-
Role of calpains in the injury-induced dysfunction and degeneration of the mammalian axon.Neurobiol Dis. 2013 Dec;60:61-79. doi: 10.1016/j.nbd.2013.08.010. Epub 2013 Aug 19. Neurobiol Dis. 2013. PMID: 23969238 Free PMC article. Review.
-
Mechanisms of axonal injury: internodal nanocomplexes and calcium deregulation.Trends Mol Med. 2010 Apr;16(4):160-70. doi: 10.1016/j.molmed.2010.02.002. Epub 2010 Mar 6. Trends Mol Med. 2010. PMID: 20207196 Free PMC article. Review.
-
Regulation of podosome formation in macrophages by a splice variant of the sodium channel SCN8A.J Biol Chem. 2009 Mar 20;284(12):8114-26. doi: 10.1074/jbc.M801892200. Epub 2009 Jan 9. J Biol Chem. 2009. PMID: 19136557 Free PMC article.
-
Axonal degeneration as a therapeutic target in the CNS.Cell Tissue Res. 2012 Jul;349(1):289-311. doi: 10.1007/s00441-012-1362-3. Epub 2012 Mar 6. Cell Tissue Res. 2012. PMID: 22392734 Free PMC article. Review.
-
Complex interplay between glutamate receptors and intracellular Ca2+ stores during ischaemia in rat spinal cord white matter.J Physiol. 2006 Nov 15;577(Pt 1):191-204. doi: 10.1113/jphysiol.2006.116798. Epub 2006 Aug 31. J Physiol. 2006. PMID: 16945971 Free PMC article.
References
-
- Allard B, Rougier O (1992) Reappraisal of the role of sodium ions in excitation-contraction coupling in frog twitch muscle. J Muscle Res Cell Motil 13: 117-125. - PubMed
-
- Arundine M, Tymianski M (2003) Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34: 325-337. - PubMed
-
- Banasiak KJ, Burenkova O, Haddad GG (2004) Activation of voltage-sensitive sodium channels during oxygen deprivation leads to apoptotic neuronal death. Neuroscience 126: 31-44. - PubMed
-
- Bernardi P (1999) Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev 79: 1127-1155. - PubMed
-
- Berridge MJ (1998) Neuronal calcium signaling. Neuron 21: 13-26. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous