

Measures of human population structure show heterogeneity among genomic regions
- PMID: 16251456
- PMCID: PMC1310634
- DOI: 10.1101/gr.4398405
Measures of human population structure show heterogeneity among genomic regions
Abstract
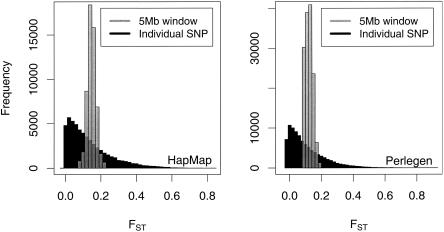
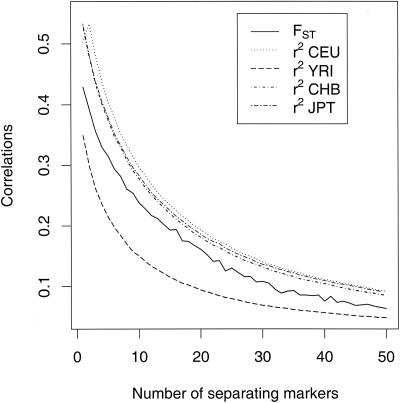
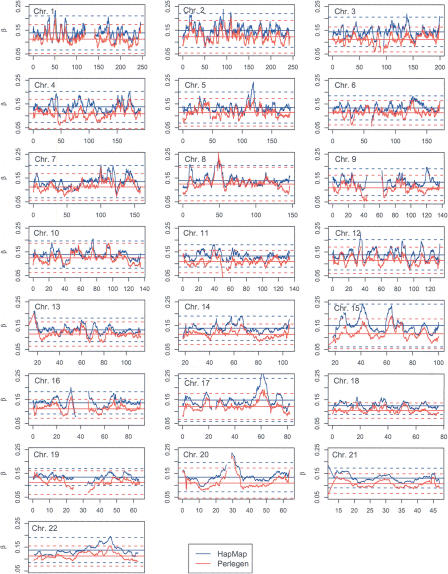
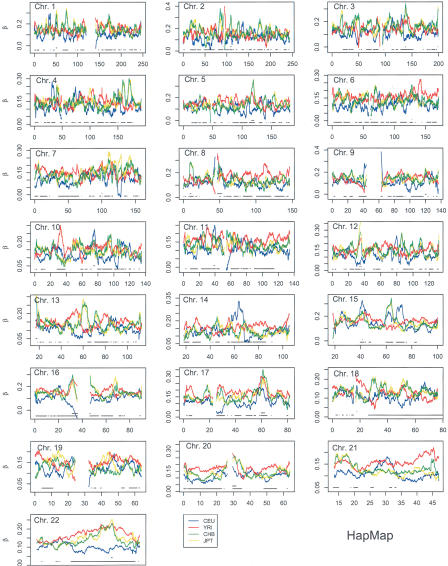
Estimates of genetic population structure (F(ST)) were constructed from all autosomes in two large SNP data sets. The Perlegen data set contains genotypes on approximately 1 million SNPs segregating in all three samples of Americans of African, Asian, and European descent; and the Phase I HapMap data set contains genotypes on approximately 0.6 million SNPs segregating in all four samples from specific Caucasian, Chinese, Japanese, and Yoruba populations. Substantial heterogeneity of F(ST) values was found between segments within chromosomes, although there was similarity between the two data sets. There was also substantial heterogeneity among population-specific F(ST) values, with the relative sizes of these values often changing along each chromosome. Population-structure estimates are often used as indicators of natural selection, but the analyses presented here show that individual-marker estimates are too variable to be useful. There is inherent variation in these statistics because of variation in genealogy even among neutral loci, and values at pairs of loci are correlated to an extent that reflects the linkage disequilibrium between them. Furthermore, it may be that the best indications of selection will come from population-specific F(ST) values rather than the usually reported population-average values.
Figures
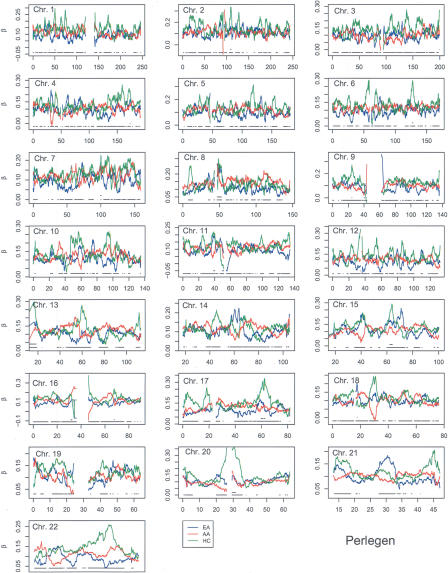
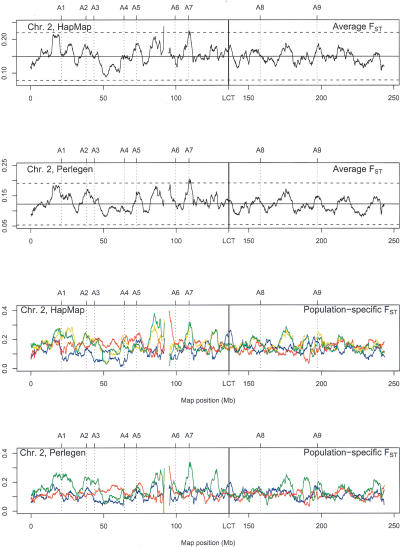
References
-
- Cockerham, C.C. and Weir, B.S. 1983. Variance of actual inbreeding. Theor. Popul. Biol. 23: 85–109. - PubMed
-
- Dodds, K.G. 1986. “Resampling methods in genetics and the effect of family structure in genetic data.” Ph.D. thesis, North Carolina State University, Raleigh.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous