

Ascertainment bias in studies of human genome-wide polymorphism
- PMID: 16251459
- PMCID: PMC1310637
- DOI: 10.1101/gr.4107905
Ascertainment bias in studies of human genome-wide polymorphism
Abstract
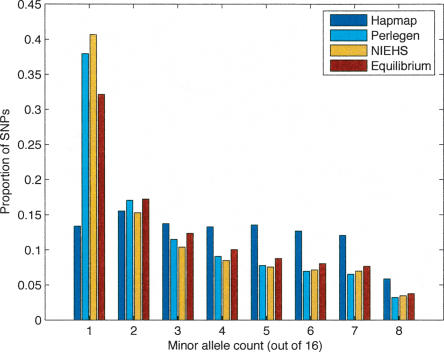
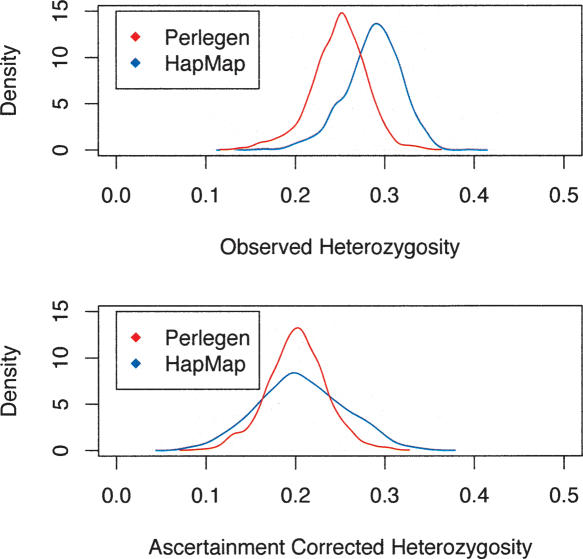
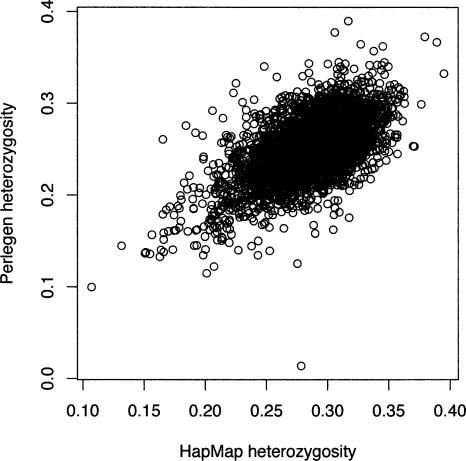
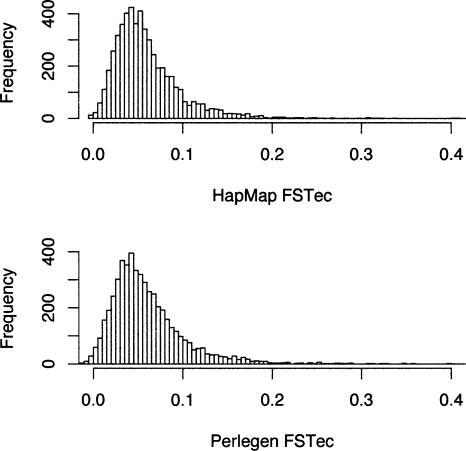
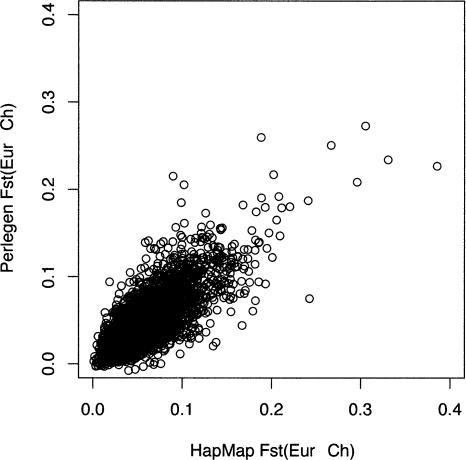
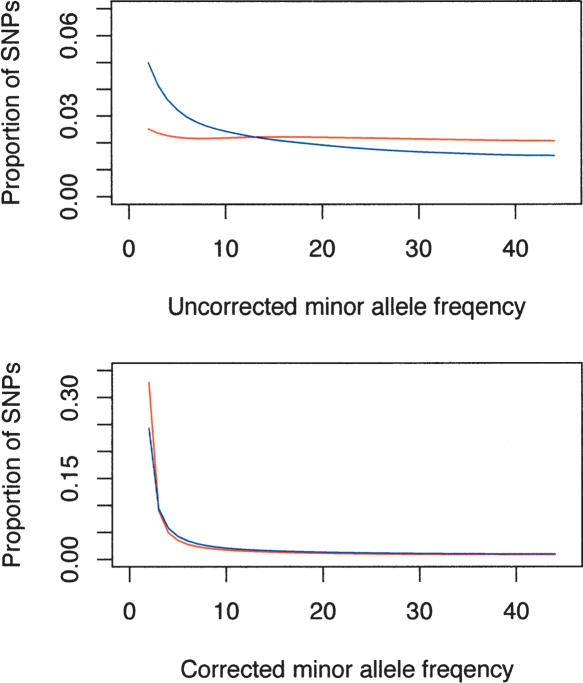
Large-scale SNP genotyping studies rely on an initial assessment of nucleotide variation to identify sites in the DNA sequence that harbor variation among individuals. This "SNP discovery" sample may be quite variable in size and composition, and it has been well established that properties of the SNPs that are found are influenced by the discovery sampling effort. The International HapMap project relied on nearly any piece of information available to identify SNPs-including BAC end sequences, shotgun reads, and differences between public and private sequences-and even made use of chimpanzee data to confirm human sequence differences. In addition, the ascertainment criteria shifted from using only SNPs that had been validated in population samples, to double-hit SNPs, to finally accepting SNPs that were singletons in small discovery samples. In contrast, Perlegen's primary discovery was a resequencing-by-hybridization effort using the 24 people of diverse origin in the Polymorphism Discovery Resource. Here we take these two data sets and contrast two basic summary statistics, heterozygosity and F(ST), as well as the site frequency spectra, for 500-kb windows spanning the genome. The magnitude of disparity between these samples in these measures of variability indicates that population genetic analysis on the raw genotype data is ill advised. Given the knowledge of the discovery samples, we perform an ascertainment correction and show how the post-correction data are more consistent across these studies. However, discrepancies persist, suggesting that the heterogeneity in the SNP discovery process of the HapMap project resulted in a data set resistant to complete ascertainment correction. Ascertainment bias will likely erode the power of tests of association between SNPs and complex disorders, but the effect will likely be small, and perhaps more importantly, it is unlikely that the bias will introduce false-positive inferences.
Figures
References
-
- Akey, J.M., Zhang, K., Xiong, M., and Jin, L. 2003. The effect of single nucleotide polymorphism identification strategies on estimates of linkage disequilibrium. Mol. Biol. Evol. 20: 232–242. - PubMed
-
- Bustamante, C.D., Fledel-Alon, A., Williamson, S., Nielsen, R., Hubisz, M.T., Glanowski, S., Tanenbaum, D.M., White, T.J., Sninsky, J.J., Hernandez, R., et al. 2005. Natural selection on protein coding genes in the human genome. Nature (in press). - PubMed
-
- Crawford, D.C., Carlson, C.S., Rieder, M.J., Carrington, D.P., Yi, Q., Smith, J.D., Eberle, M.A., Kruglyak, L., and Nickerson, D.A. 2004. Haplotype diversity across 100 candidate genes for inflammation, lipid metabolism, and blood pressure regulation in two populations. Am. J. Hum. Genet. 74: 610–622. - PMC - PubMed
-
- Gibbs, R.A., Belmont, J.W., Hardenbol, P., Willis, T.D., Yu, F., Yang, H., Chang, L.-Y., Huang, W., Liu, B., Shen, Y., et al. 2003. The International HapMap Project. Nature 426: 789–796. - PubMed
Web site references
-
- http://egp.gs.washington.edu; NIEHS resequencing study.
-
- http://www.hapmap.org; International HapMap Project.
-
- http://genome.perlegen.com/browser/download.html; Perlegen Sciences Web site.
-
- http://www.hapmap.org/downloads/encode1.html; HapMap .subjects
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous