

Genomic scans for selective sweeps using SNP data
- PMID: 16251466
- PMCID: PMC1310644
- DOI: 10.1101/gr.4252305
Genomic scans for selective sweeps using SNP data
Abstract
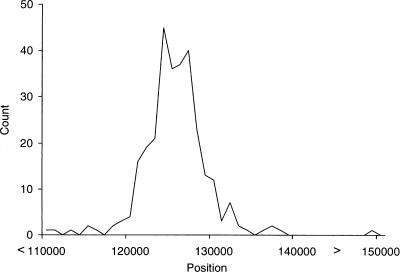
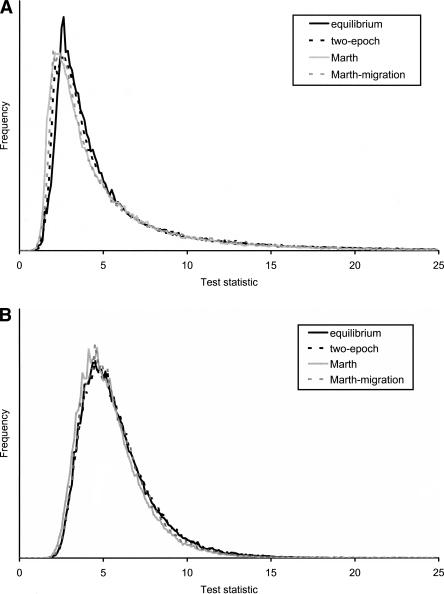
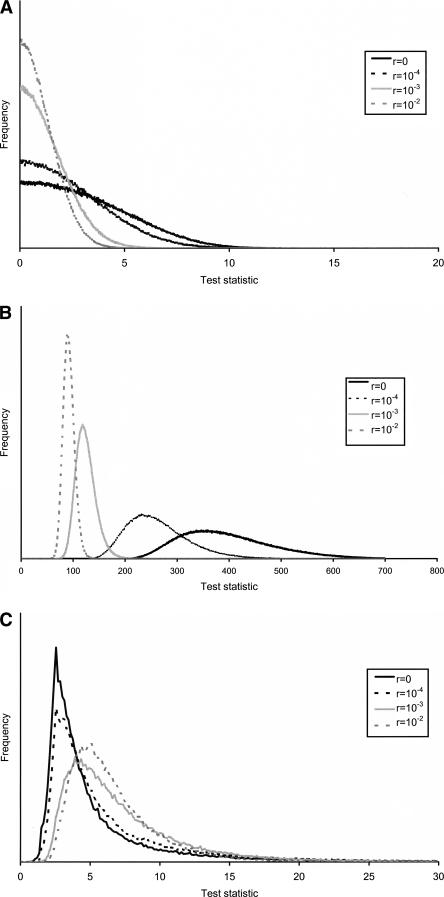
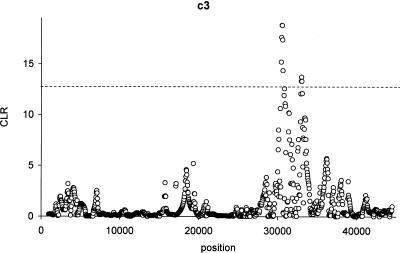
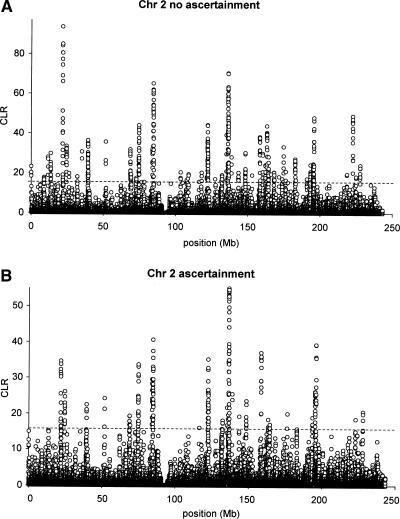
Detecting selective sweeps from genomic SNP data is complicated by the intricate ascertainment schemes used to discover SNPs, and by the confounding influence of the underlying complex demographics and varying mutation and recombination rates. Current methods for detecting selective sweeps have little or no robustness to the demographic assumptions and varying recombination rates, and provide no method for correcting for ascertainment biases. Here, we present several new tests aimed at detecting selective sweeps from genomic SNP data. Using extensive simulations, we show that a new parametric test, based on composite likelihood, has a high power to detect selective sweeps and is surprisingly robust to assumptions regarding recombination rates and demography (i.e., has low Type I error). Our new test also provides estimates of the location of the selective sweep(s) and the magnitude of the selection coefficient. To illustrate the method, we apply our approach to data from the Seattle SNP project and to Chromosome 2 data from the HapMap project. In Chromosome 2, the most extreme signal is found in the lactase gene, which previously has been shown to be undergoing positive selection. Evidence for selective sweeps is also found in many other regions, including genes known to be associated with disease risk such as DPP10 and COL4A3.
Figures
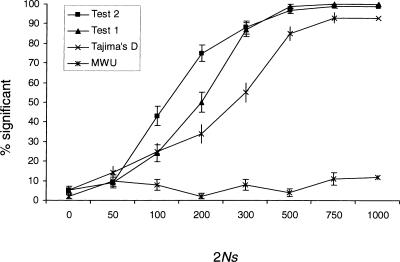
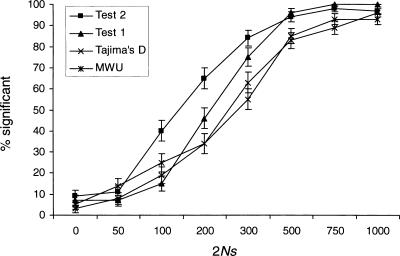
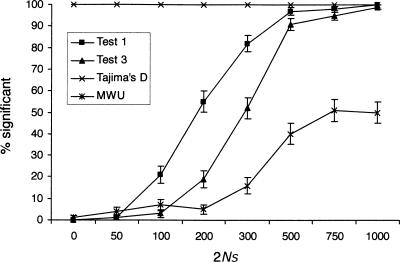
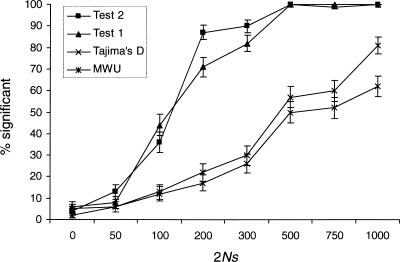
References
-
- Barton, N.H. 1998. The effect of hitch-hiking on neutral genealogies. Genet. Res. 72: 123–133.
Web site references
-
- http://pga.gs.washington.edu; the Seattle SNP database [Feb. 2004]—SeattleSNPs. NHLBI Program for Genomic Applications, SeattleSNPs, Seattle, WA.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials