

Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells
- PMID: 16252232
- PMCID: PMC1271382
- DOI: 10.1086/497348
Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells
Abstract
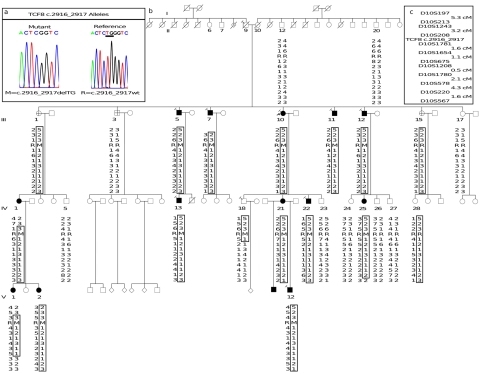
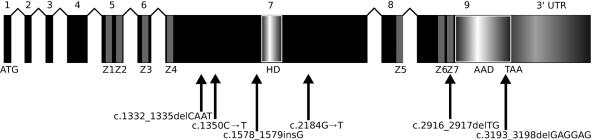
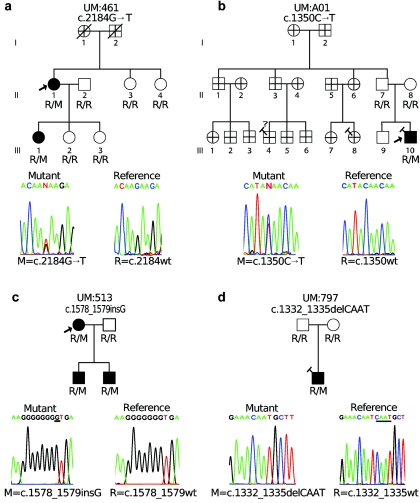
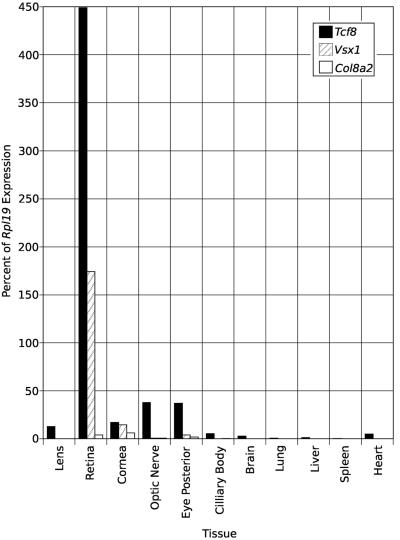
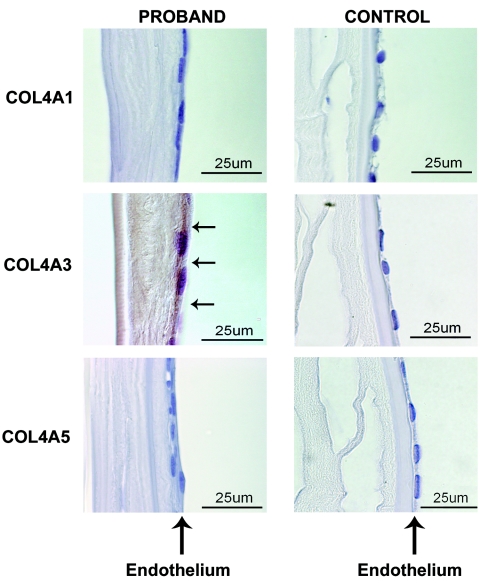
Posterior polymorphous corneal dystrophy (PPCD, also known as PPMD) is a rare disease involving metaplasia and overgrowth of corneal endothelial cells. In patients with PPCD, these cells manifest in an epithelial morphology and gene expression pattern, produce an aberrant basement membrane, and, sometimes, spread over the iris and nearby structures in a way that increases the risk for glaucoma. We previously mapped PPCD to a region (PPCD3) on chromosome 10 containing the gene that encodes the two-handed zinc-finger homeodomain transcription factor TCF8. Here, we report a heterozygous frameshift mutation in TCF8 that segregates with PPCD in the family used to map PPCD3 and four different heterozygous nonsense and frameshift mutations in TCF8 in four other PPCD probands. Family reports of inguinal hernia, hydrocele, and possible bone anomalies in affected individuals suggest that individuals with TCF8 mutations should be examined for nonocular anomalies. We detect transcripts of all three identified PPCD genes (VSX1, COL8A2, and TCF8) in the cornea. We show presence of a complex (core plus secondary) binding site for TCF8 in the promoter of Alport syndrome gene COL4A3, which encodes collagen type IV alpha 3, and we present immunohistochemical evidence of ectopic expression of COL4A3 in corneal endothelium of the proband of the original PPCD3 family. Identification of TCF8 as the PPCD3 gene provides a valuable tool for the study of critical gene regulation events in PPCD pathology and suggests a possible role for TCF8 mutations in altered structure and function of cells lining body cavities other than the anterior chamber of the eye. Thus, this study has identified TCF8 as the gene responsible for approximately half of the cases of PPCD, has implicated TCF8 mutations in developmental abnormalities outside the eye, and has presented the TCF8 regulatory target, COL4A3, as a key, shared molecular component of two different diseases, PPCD and Alport syndrome.
Figures
Similar articles
-
Posterior polymorphous corneal dystrophy is associated with TCF8 gene mutations and abdominal hernia.Am J Med Genet A. 2007 Nov 1;143A(21):2549-56. doi: 10.1002/ajmg.a.31978. Am J Med Genet A. 2007. PMID: 17935237
-
Analysis of the role of ZEB1 in the pathogenesis of posterior polymorphous corneal dystrophy.Invest Ophthalmol Vis Sci. 2012 Jan 25;53(1):273-8. doi: 10.1167/iovs.11-8038. Invest Ophthalmol Vis Sci. 2012. PMID: 22199242 Free PMC article.
-
Mutational spectrum of the ZEB1 gene in corneal dystrophies supports a genotype-phenotype correlation.Invest Ophthalmol Vis Sci. 2013 May 3;54(5):3215-23. doi: 10.1167/iovs.13-11781. Invest Ophthalmol Vis Sci. 2013. PMID: 23599324
-
Genetics of the corneal endothelial dystrophies: an evidence-based review.Clin Genet. 2013 Aug;84(2):109-19. doi: 10.1111/cge.12191. Epub 2013 Jun 10. Clin Genet. 2013. PMID: 23662738 Free PMC article. Review.
-
Update on the genetics of corneal endothelial dystrophies.Indian J Ophthalmol. 2022 Jul;70(7):2239-2248. doi: 10.4103/ijo.IJO_992_22. Indian J Ophthalmol. 2022. PMID: 35791103 Free PMC article. Review.
Cited by
-
Differing roles for TCF4 and COL8A2 in central corneal thickness and fuchs endothelial corneal dystrophy.PLoS One. 2012;7(10):e46742. doi: 10.1371/journal.pone.0046742. Epub 2012 Oct 23. PLoS One. 2012. PMID: 23110055 Free PMC article.
-
Snail Track Lesion with Flat Keratometry in Anterior Segment Dysgenesis Caused by a Novel FOXC1 Variant.J Clin Med. 2022 Aug 31;11(17):5166. doi: 10.3390/jcm11175166. J Clin Med. 2022. PMID: 36079096 Free PMC article.
-
Corneal dystrophies.Orphanet J Rare Dis. 2009 Feb 23;4:7. doi: 10.1186/1750-1172-4-7. Orphanet J Rare Dis. 2009. PMID: 19236704 Free PMC article. Review.
-
Genetic analysis of chromosome 20-related posterior polymorphous corneal dystrophy: genetic heterogeneity and exclusion of three candidate genes.Mol Vis. 2008 Jan 16;14:71-80. Mol Vis. 2008. PMID: 18253095 Free PMC article.
-
Transcriptomic Profiling of Posterior Polymorphous Corneal Dystrophy.Invest Ophthalmol Vis Sci. 2017 Jun 1;58(7):3202-3214. doi: 10.1167/iovs.17-21423. Invest Ophthalmol Vis Sci. 2017. PMID: 28654985 Free PMC article.
References
Web Resources
-
- BodyMap Database, http://bodymap.ims.u-tokyo.ac.jp/
-
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/ (for c.685A→G [accession number rs220060])
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for VSX1 [accession number NM_199425], COL8A2 [accession number NM_005202], TCF8 [accession number NM_030751], Tcf8 [accession number NM_011546], Vsx1 [accession number NM_054068], Col8a2 [accession number NM_199473], Rpl19 [accession number NM_009078])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
-
- The Sequence Manipulation Suite, http://bio.ifom-firc.it/TOOLS/sms/main.html/
References
-
- Bachoo S, Gibbons RJ (1999) Germline and gonosomal mosaicism in the ATR-X syndrome. Eur J Hum Genet 7:933–936 - PubMed
-
- Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, Gerken SC, Gregory MC, Skolnick MH, Atkin CL, Tryggvason K (1990) Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248:1224–1227 - PubMed
-
- Beck JA, Poulter M, Campbell TA, Uphill JB, Adamson G, Geddes JF, Revesz T, Davis MB, Wood NW, Collinge J, Tabrizi SJ (2004) Somatic and germline mosaicism in sporadic early-onset Alzheimer’s disease. Hum Mol Genet 13:1219–1224 - PubMed
-
- Bendavid R (2004) The unified theory of hernia formation. Hernia 8:171–176 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
