

Evidence for widespread reticulate evolution within human duplicons
- PMID: 16252241
- PMCID: PMC1271390
- DOI: 10.1086/497704
Evidence for widespread reticulate evolution within human duplicons
Abstract
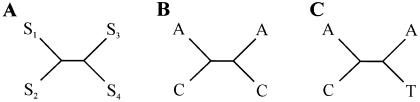
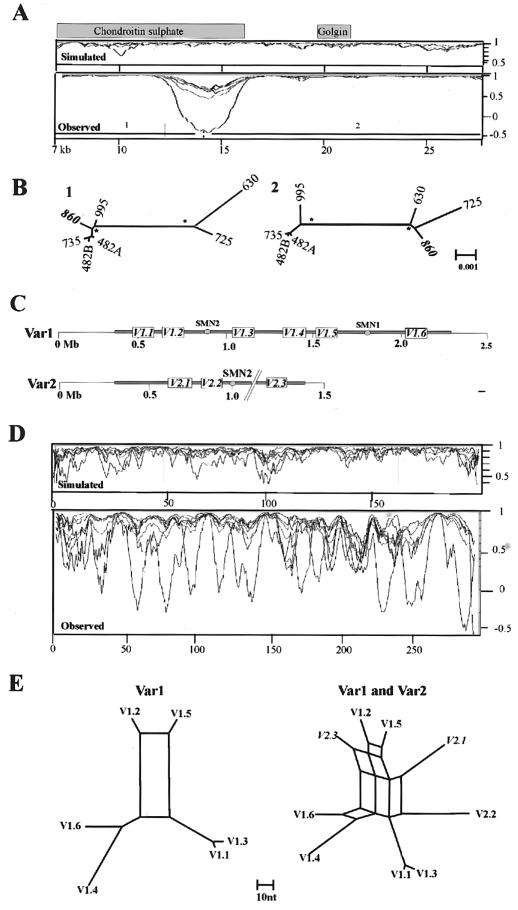
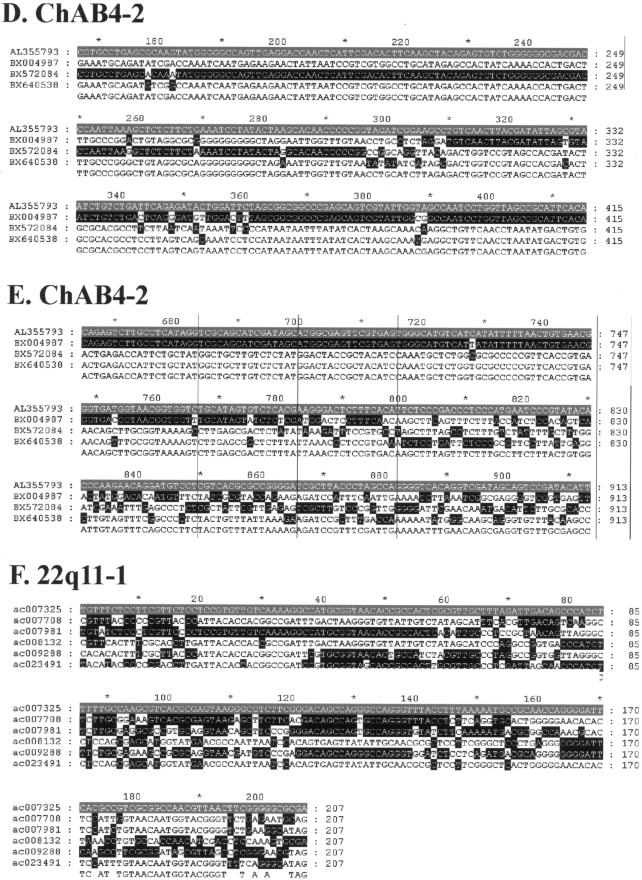
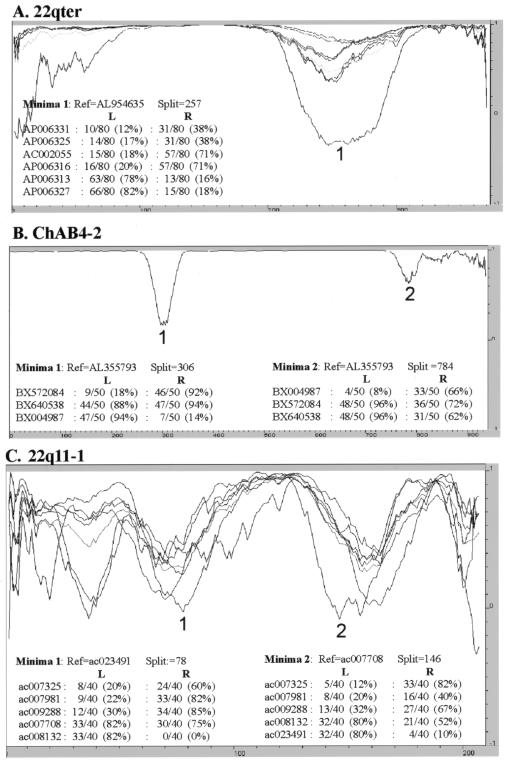
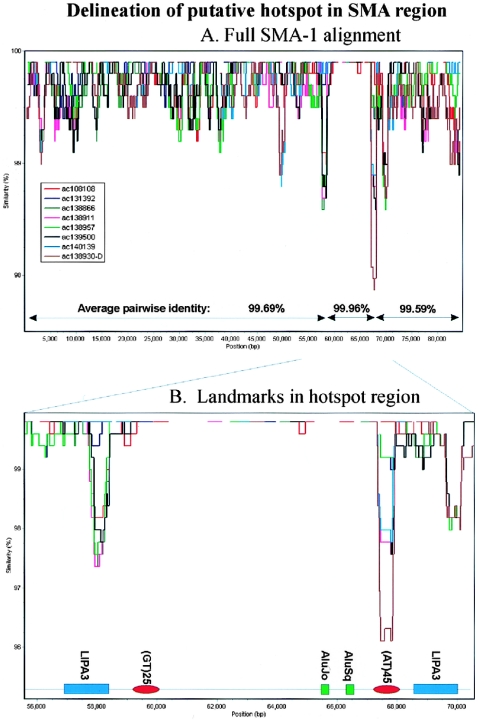
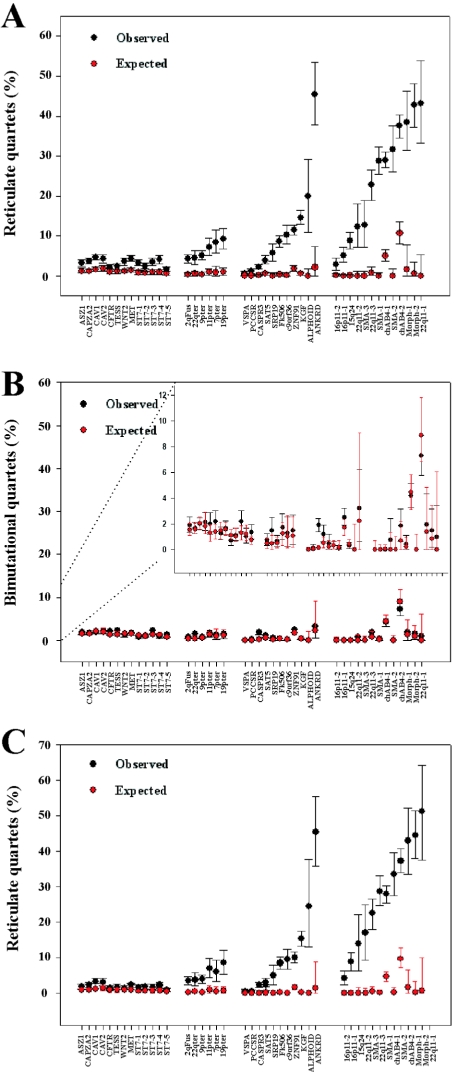
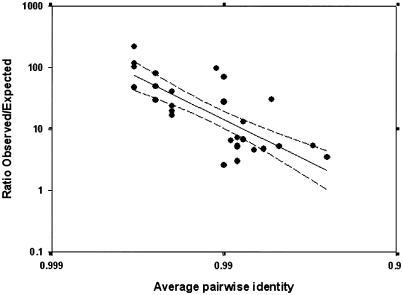
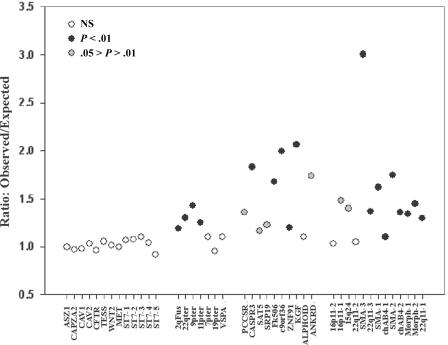
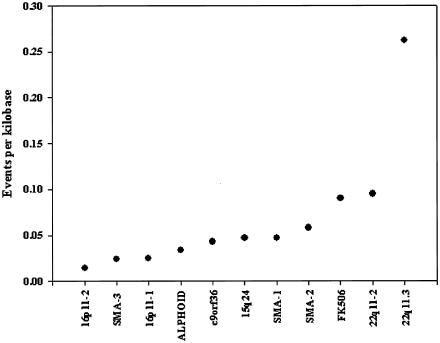
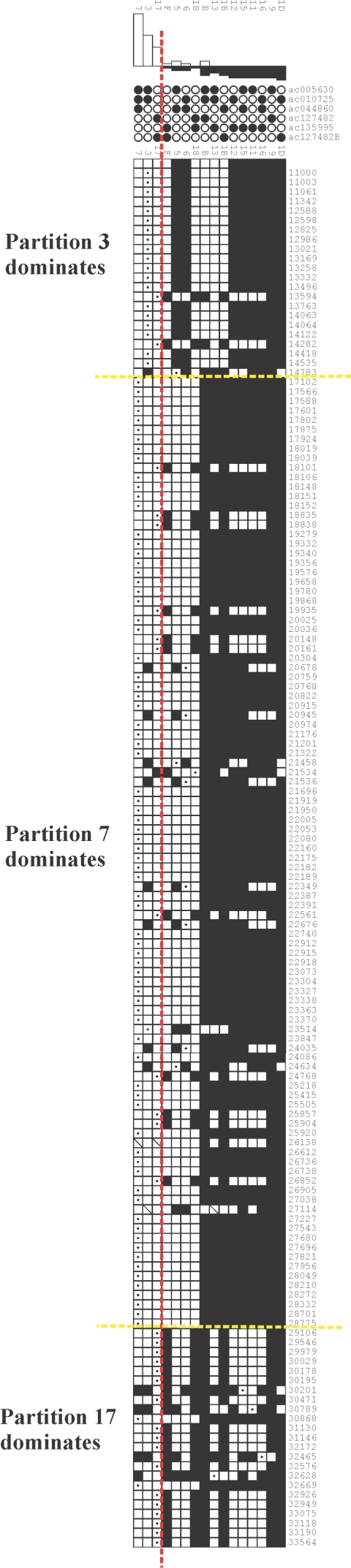
Approximately 5% of the human genome consists of segmental duplications that can cause genomic mutations and may play a role in gene innovation. Reticulate evolutionary processes, such as unequal crossing-over and gene conversion, are known to occur within specific duplicon families, but the broader contribution of these processes to the evolution of human duplications remains poorly characterized. Here, we use phylogenetic profiling to analyze multiple alignments of 24 human duplicon families that span >8 Mb of DNA. Our results indicate that none of them are evolving independently, with all alignments showing sharp discontinuities in phylogenetic signal consistent with reticulation. To analyze these results in more detail, we have developed a quartet method that estimates the relative contribution of nucleotide substitution and reticulate processes to sequence evolution. Our data indicate that most of the duplications show a highly significant excess of sites consistent with reticulate evolution, compared with the number expected by nucleotide substitution alone, with 15 of 30 alignments showing a >20-fold excess over that expected. Using permutation tests, we also show that at least 5% of the total sequence shares 100% sequence identity because of reticulation, a figure that includes 74 independent tracts of perfect identity >2 kb in length. Furthermore, analysis of a subset of alignments indicates that the density of reticulation events is as high as 1 every 4 kb. These results indicate that phylogenetic relationships within recently duplicated human DNA can be rapidly disrupted by reticulate evolution. This finding has important implications for efforts to finish the human genome sequence, complicates comparative sequence analysis of duplicon families, and could profoundly influence the tempo of gene-family evolution.
Figures


References
Web Resources
-
- Algorithms in Bioinformatics: SplitsTree4, http://www-ab.informatik.uni-tuebingen.de/software/splits/welcome.html (for D. H. Huson and D. Bryant's work on estimating phylogenetic trees and networks using SplitsTree4)
-
- NISC Comparative Vertebrate Sequencing, http://www.nisc.nih.gov/open_page.html?/projects/comp_seq.html (for Target 1)
-
- Pairwise FLAG, http://bioinformatics.itri.org.tw/prflag/prflag.php
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410 - PubMed
-
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE (2002) Recent segmental duplications in the human genome. Science 297:1003–1007 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
