

A haplotype map of the human genome
- PMID: 16255080
- PMCID: PMC1880871
- DOI: 10.1038/nature04226
A haplotype map of the human genome
Abstract
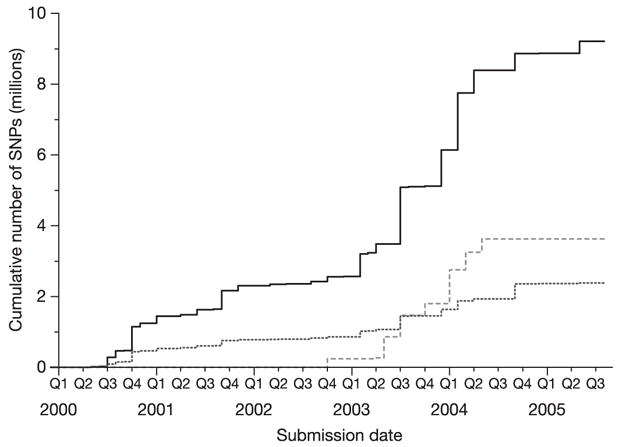
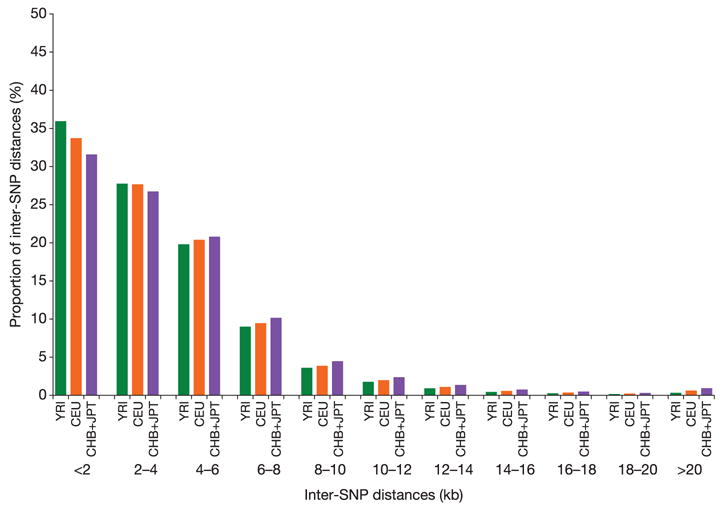
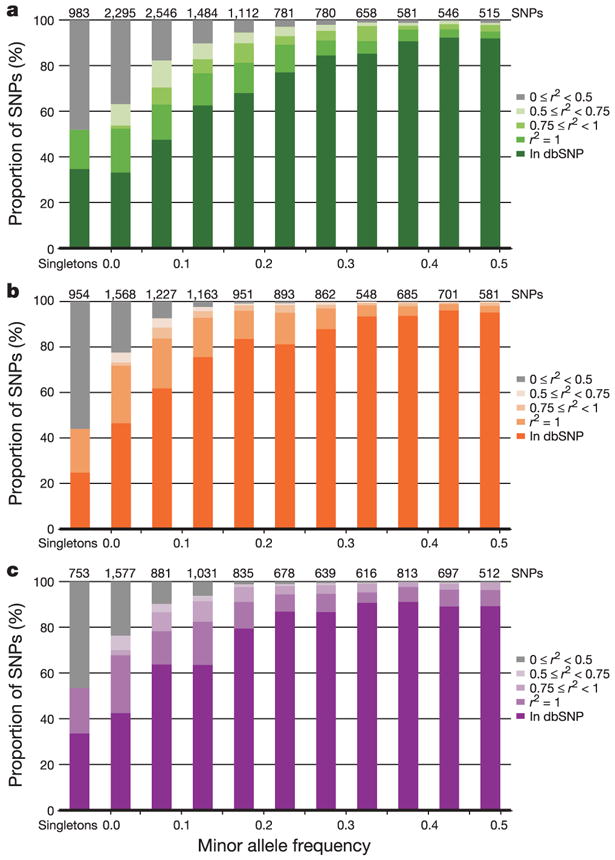
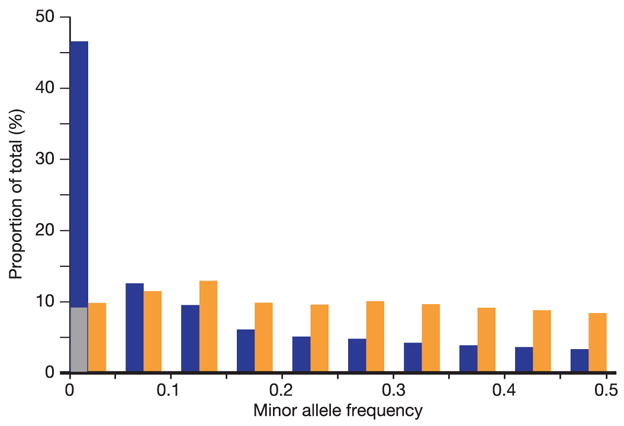
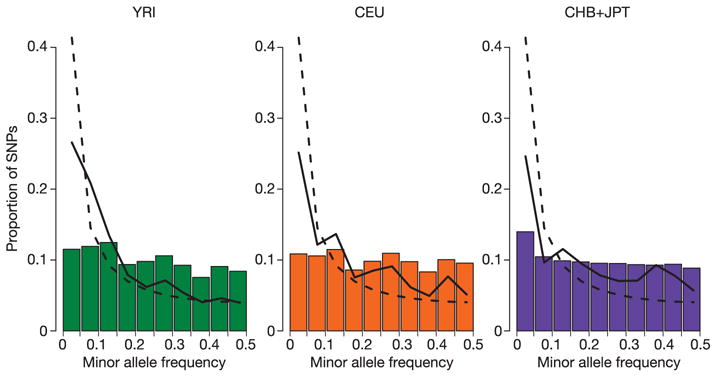
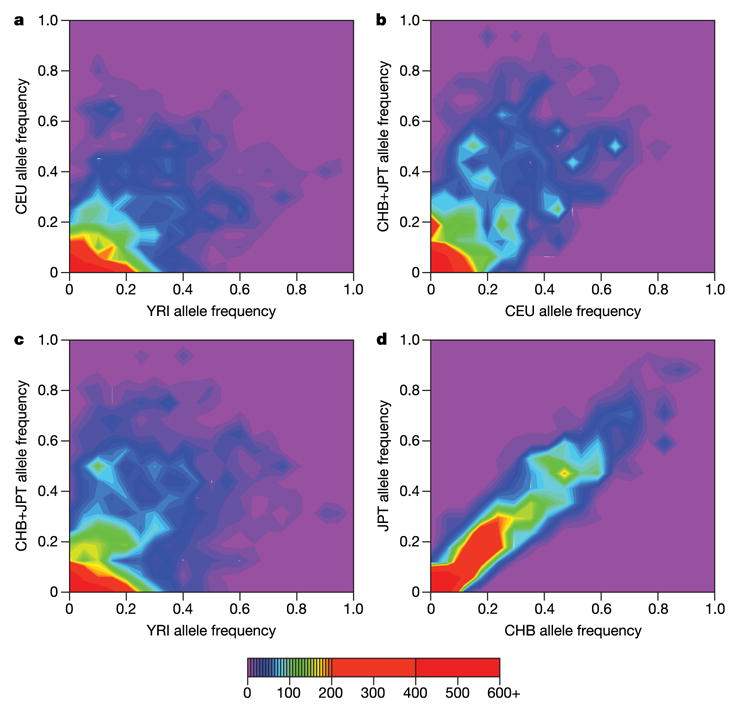
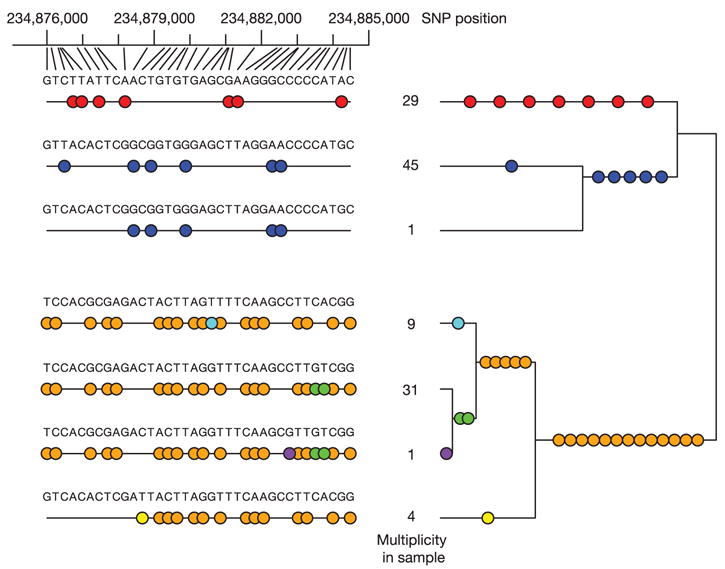
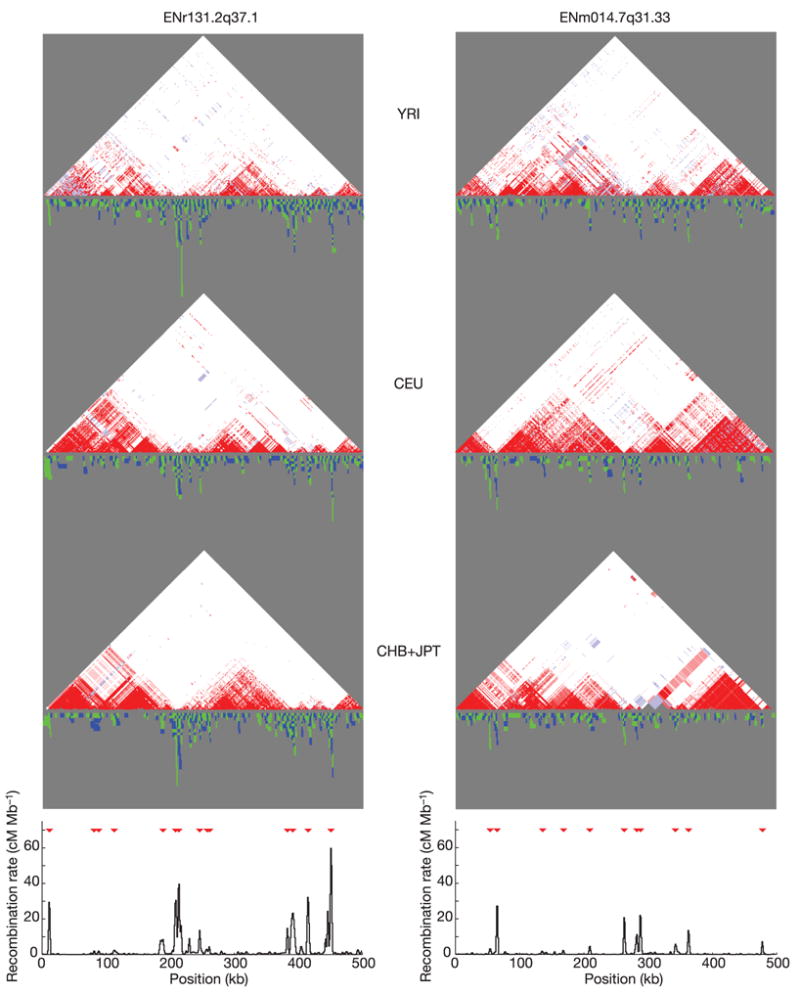
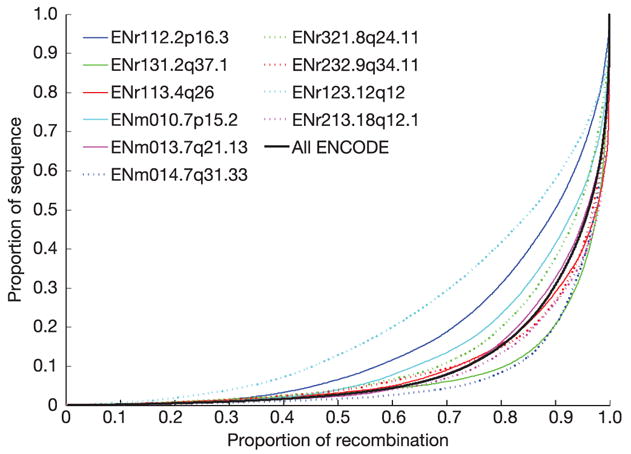
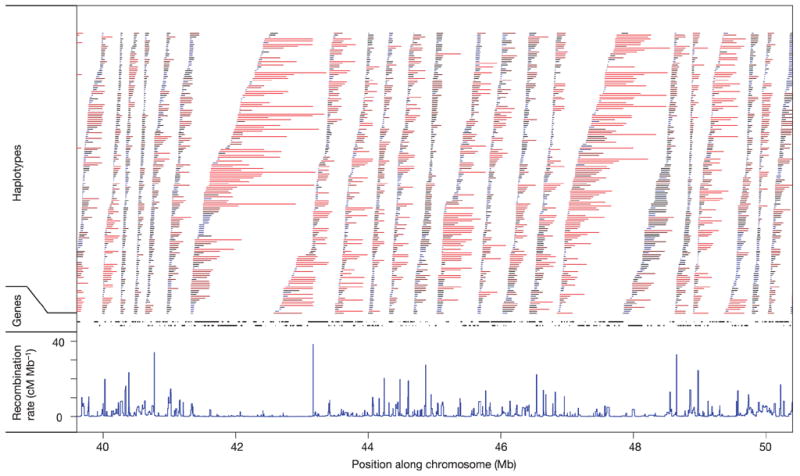
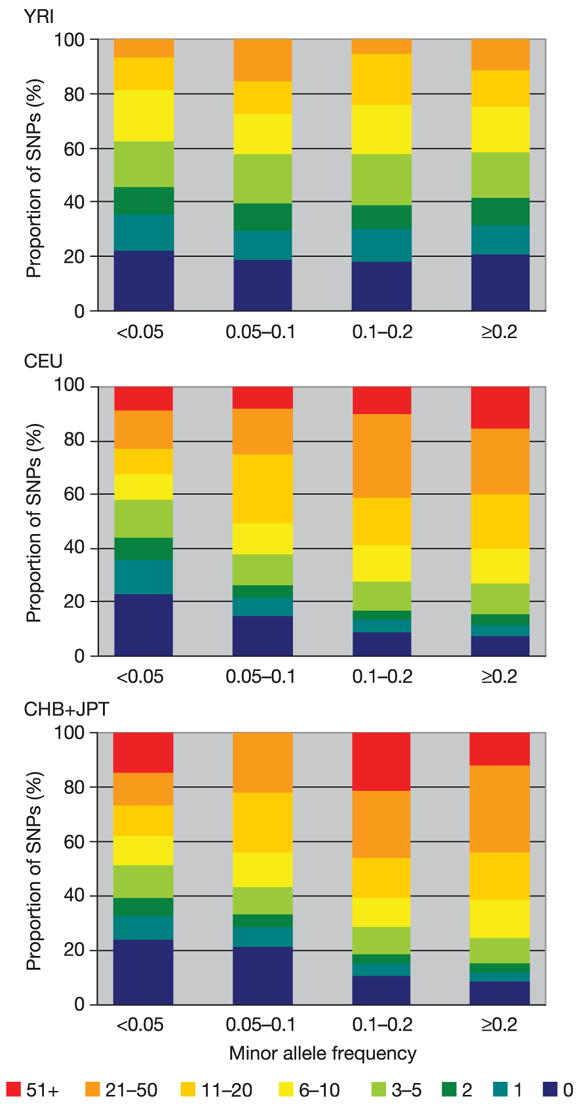
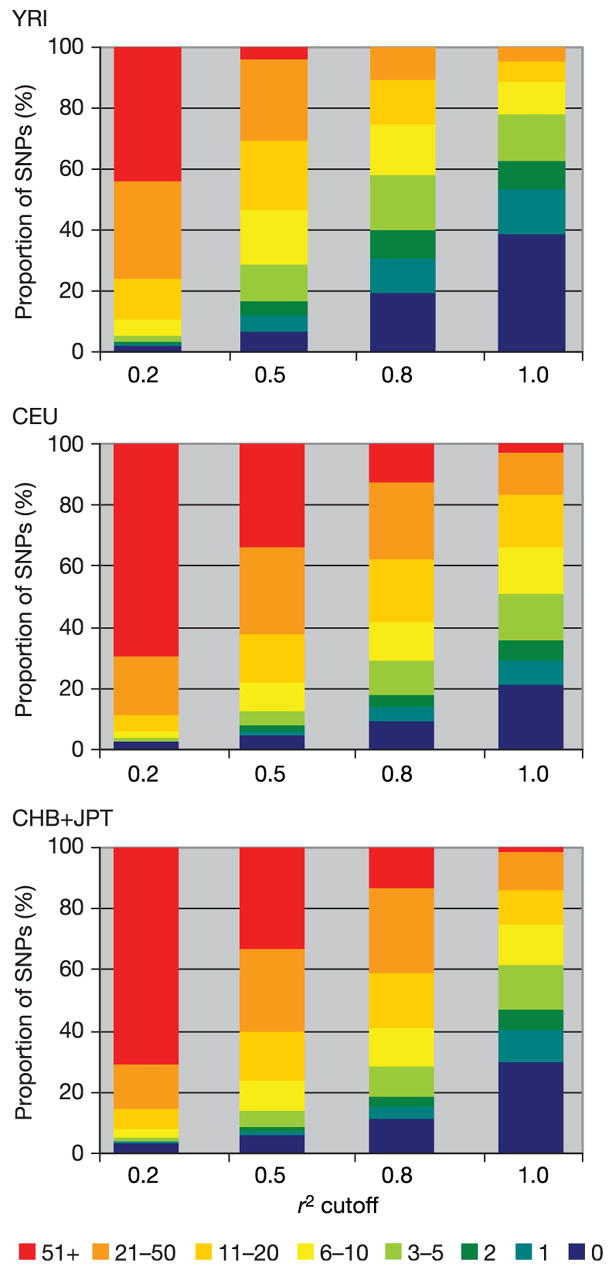
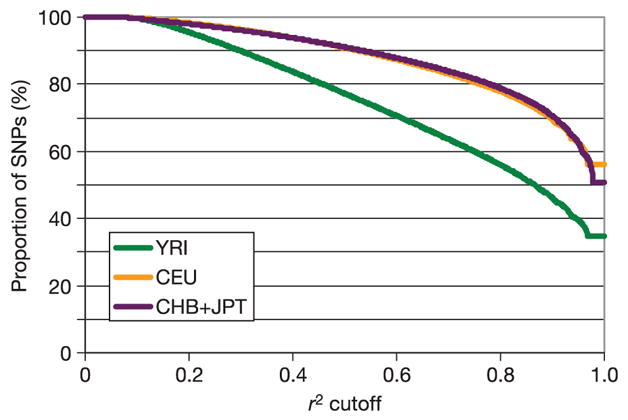
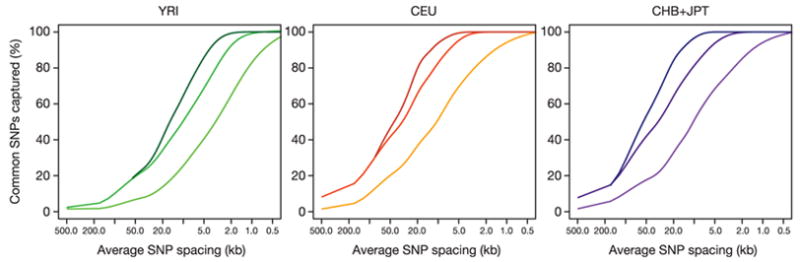
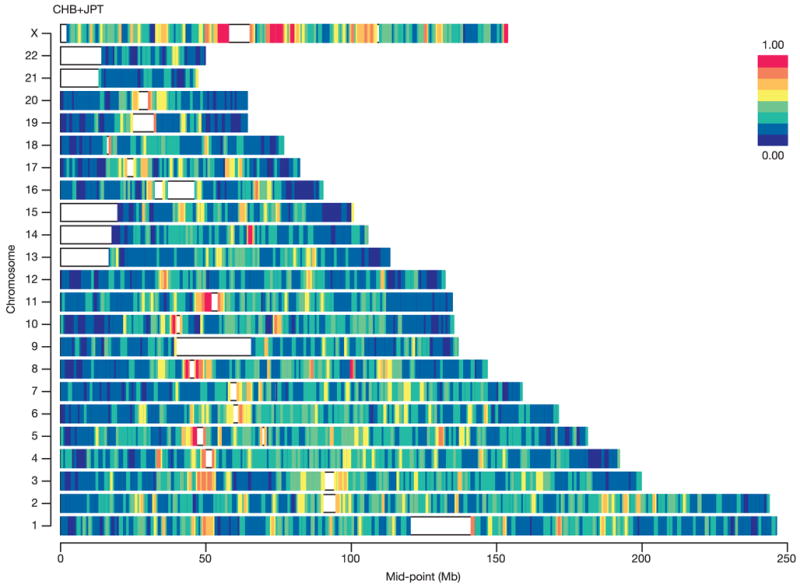
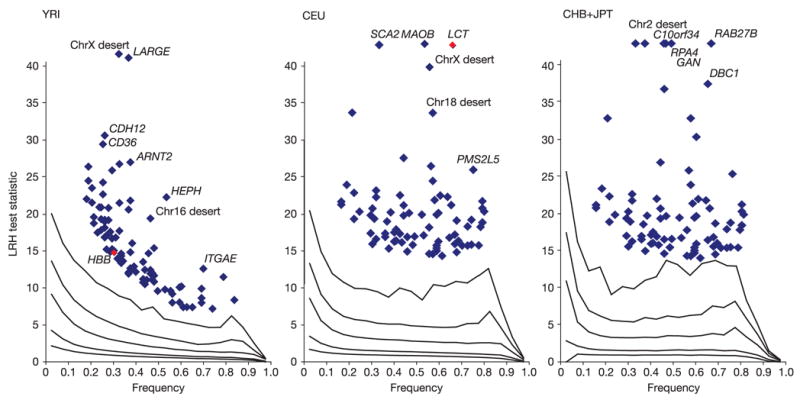
Inherited genetic variation has a critical but as yet largely uncharacterized role in human disease. Here we report a public database of common variation in the human genome: more than one million single nucleotide polymorphisms (SNPs) for which accurate and complete genotypes have been obtained in 269 DNA samples from four populations, including ten 500-kilobase regions in which essentially all information about common DNA variation has been extracted. These data document the generality of recombination hotspots, a block-like structure of linkage disequilibrium and low haplotype diversity, leading to substantial correlations of SNPs with many of their neighbours. We show how the HapMap resource can guide the design and analysis of genetic association studies, shed light on structural variation and recombination, and identify loci that may have been subject to natural selection during human evolution.
Figures
Comment in
-
Genomics: understanding human diversity.Nature. 2005 Oct 27;437(7063):1241-2. doi: 10.1038/4371241a. Nature. 2005. PMID: 16251937 No abstract available.
References
-
- Lechler R, Warrens A. HLA in Health and Disease. 2. Academic Press; San Diego, California: 2005.
-
- Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer’s disease. Annu Rev Neurosci. 1996;19:53–77. - PubMed
-
- Dahlbäck B. Resistance to activated protein C caused by the factor V R506Q mutation is a common risk factor for venous thrombosis. Thromb Haemost. 1997;78:483–488. - PubMed
-
- Altshuler D, et al. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nature Genet. 2000;26:76–80. - PubMed
-
- Deeb SS, et al. A Pro12Ala substitution in PPARγ2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nature Genet. 1998;20:284–287. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
