

Antagonists of Hsp16.3, a low-molecular-weight mycobacterial chaperone and virulence factor, derived from phage-displayed peptide libraries
- PMID: 16269776
- PMCID: PMC1287729
- DOI: 10.1128/AEM.71.11.7334-7344.2005
Antagonists of Hsp16.3, a low-molecular-weight mycobacterial chaperone and virulence factor, derived from phage-displayed peptide libraries
Abstract
The persistence of Mycobacterium tuberculosis is a major cause of concern in tuberculosis (TB) therapy. In the persistent mode the pathogen can resist drug therapy, allowing the possibility of reactivation of the disease. Several protein factors have been identified that contribute to persistence, one of them being the 16-kDa low-molecular-weight mycobacterial heat shock protein Hsp16.3, a homologue of the mammalian eye lens protein alpha-crystallin. It is believed that Hsp16.3 plays a key role in the persistence phase by protecting essential proteins from being irreversibly denatured. Because of the close association of Hsp16.3 with persistence, an attempt has been made to develop inhibitors against it. Random peptide libraries displayed on bacteriophage M13 were screened for Hsp16.3 binding. Two phage clones were identified that bind to the Hsp16.3 protein. The corresponding synthetic peptides, an 11-mer and a 16-mer, were able to bind Hsp16.3 and inhibit its chaperone activity in vitro in a dose-dependent manner. Little or no effect of these peptides was observed on alphaB-crystallin, a homologous protein that is a key component of human eye lens, indicating that there is an element of specificity in the observed inhibition. Two histidine residues appear to be common to the selected peptides. Nuclear magnetic resonance studies performed with the 11-mer peptide indicate that in this case these two histidines may be the crucial binding determinants. The peptide inhibitors of Hsp16.3 thus obtained could serve as the basis for developing potent drugs against persistent TB.
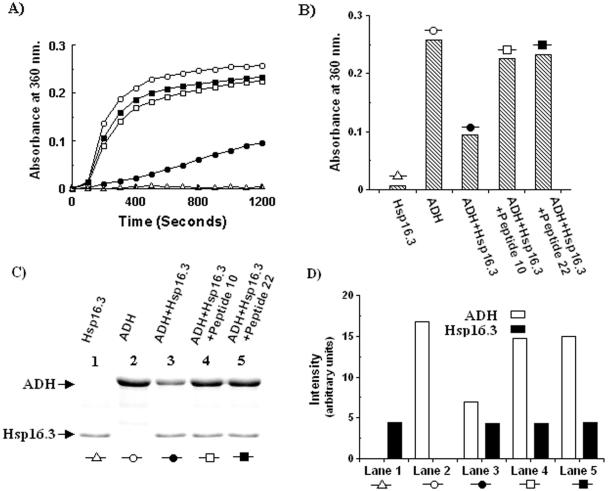
Figures





References
-
- Andreu, D., and L. Rivas. 1998. Animal antimicrobial peptides: an overview. Biopolymers 47:415-433. - PubMed
-
- Appeldoorn, C. C., T. J. Molenaar, A. Bonnefoy, S. H. van Leeuwen, P. A. Vandervoort, M. F. Hoylaerts, T. J. van Berkel, and E. A. Biessen. 2003. Rational optimization of a short human P-selectin-binding peptide leads to nanomolar affinity antagonists. J. Biol. Chem. 278:10201-10207. - PubMed
-
- Barry, C., III. November2002. Method of attenuating pathogenic mycobacteria and strains of mycobacteria so attenuated. U.S. patent 6,403,100.
-
- Bhattacharyya, T., A. Bhattacharyya, and S. Roy. 1991. A fluorescence spectroscopic study of glutaminyl-tRNA synthetase from Escherichia coli and its implications for the enzyme mechanism. Eur. J. Biochem. 200:739-745. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
