

Synaptic activity becomes excitotoxic in neurons exposed to elevated levels of platelet-activating factor
- PMID: 16276420
- PMCID: PMC1265873
- DOI: 10.1172/JCI25444
Synaptic activity becomes excitotoxic in neurons exposed to elevated levels of platelet-activating factor
Abstract
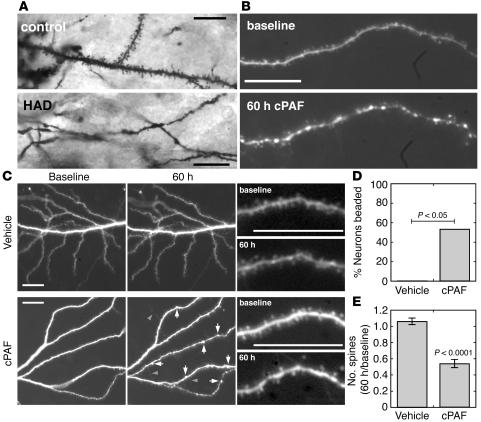
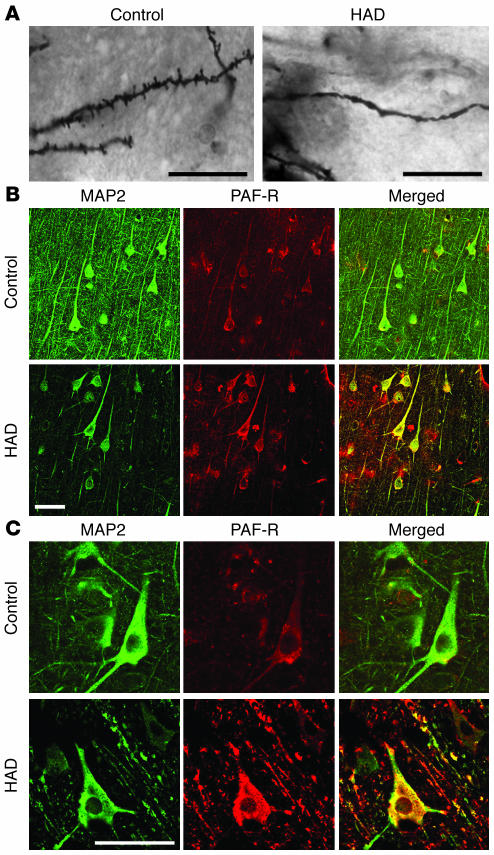
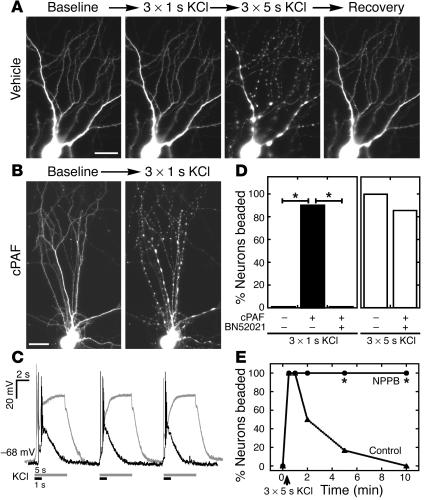
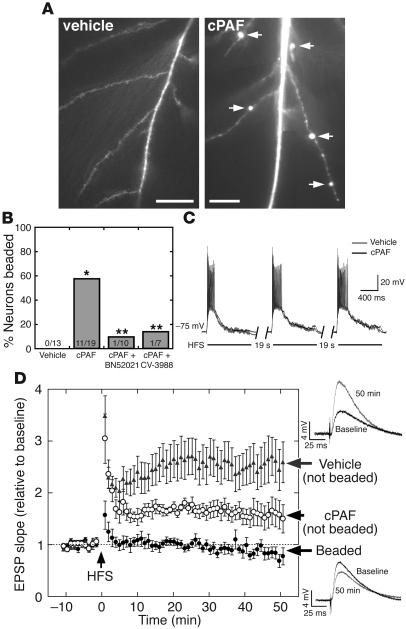
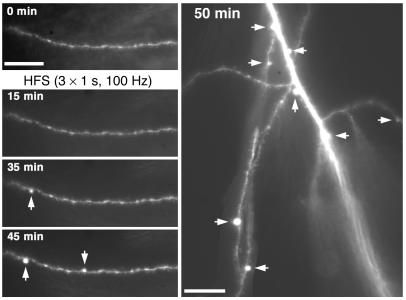
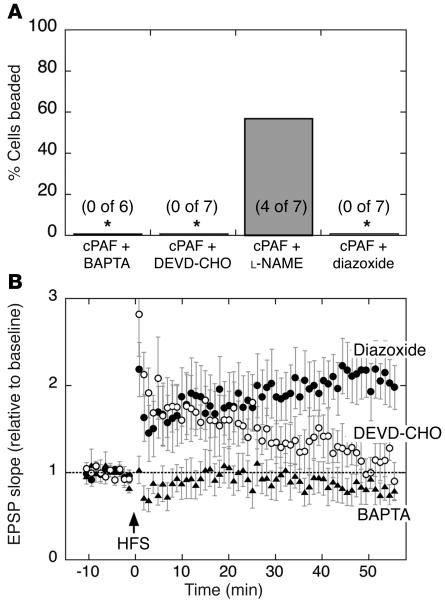
Neurologic impairment in HIV-1-associated dementia (HAD) and other neuroinflammatory diseases correlates with injury to dendrites and synapses, but how such injury occurs is not known. We hypothesized that neuroinflammation makes dendrites susceptible to excitotoxic injury following synaptic activity. We report that platelet-activating factor, an inflammatory phospholipid that mediates synaptic plasticity and neurotoxicity and is dramatically elevated in the brain during HAD, promotes dendrite injury following elevated synaptic activity and can replicate HIV-1-associated dendritic pathology. In hippocampal slices exposed to a stable platelet-activating factor analogue, tetanic stimulation that normally induces long-term synaptic potentiation instead promoted development of calcium- and caspase-dependent dendritic beading. Chemical preconditioning with diazoxide, a mitochondrial ATP-sensitive potassium channel agonist, prevented dendritic beading and restored long-term potentiation. In contrast to models invoking excessive glutamate release, these results suggest that physiologic synaptic activity may trigger excitotoxic dendritic injury during chronic neuroinflammation. Furthermore, preconditioning may represent a novel therapeutic strategy for preventing excitotoxic injury while preserving physiologic plasticity.
Figures
References
-
- Masliah E, et al. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann. Neurol. 1997;42:963–972. - PubMed
-
- Everall IP, Glass JD, McArthur J, Spargo E, Lantos P. Neuronal density in the superior frontal and temporal gyri does not correlate with the degree of human immunodeficiency virus-associated dementia. Acta Neuropathol. (Berl.). 1994;88:538–544. - PubMed
-
- Adle-Biassette H, et al. Neuronal apoptosis does not correlate with dementia in HIV infection but is related to microglial activation and axonal damage. Neuropathol. Appl. Neurobiol. 1999;25:123–133. - PubMed
-
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 1990;27:457–464. - PubMed
-
- Terry RD, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991;30:572–580. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- GM07356/GM/NIGMS NIH HHS/United States
- T32 AI049815/AI/NIAID NIH HHS/United States
- P50 MH045294/MH/NIMH NIH HHS/United States
- P01 NS031492/NS/NINDS NIH HHS/United States
- T32 GM007356/GM/NIGMS NIH HHS/United States
- R24 MH059745/MH/NIMH NIH HHS/United States
- R01 MH062962/MH/NIMH NIH HHS/United States
- MH64570/MH/NIMH NIH HHS/United States
- MH56838/MH/NIMH NIH HHS/United States
- P01 MH064570/MH/NIMH NIH HHS/United States
- MH59745/MH/NIMH NIH HHS/United States
- NS31492/NS/NINDS NIH HHS/United States
- T32-AI49815/AI/NIAID NIH HHS/United States
- MH62962/MH/NIMH NIH HHS/United States
- R01 MH056838/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
