

Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease
- PMID: 16279840
- PMCID: PMC1283364
- DOI: 10.1371/journal.pmed.0020355
Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease
Abstract
Background: The proteases (secretases) that cleave amyloid-beta (Abeta) peptide from the amyloid precursor protein (APP) have been the focus of considerable investigation in the development of treatments for Alzheimer disease. The prediction has been that reducing Abeta production in the brain, even after the onset of clinical symptoms and the development of associated pathology, will facilitate the repair of damaged tissue and removal of amyloid lesions. However, no long-term studies using animal models of amyloid pathology have yet been performed to test this hypothesis.
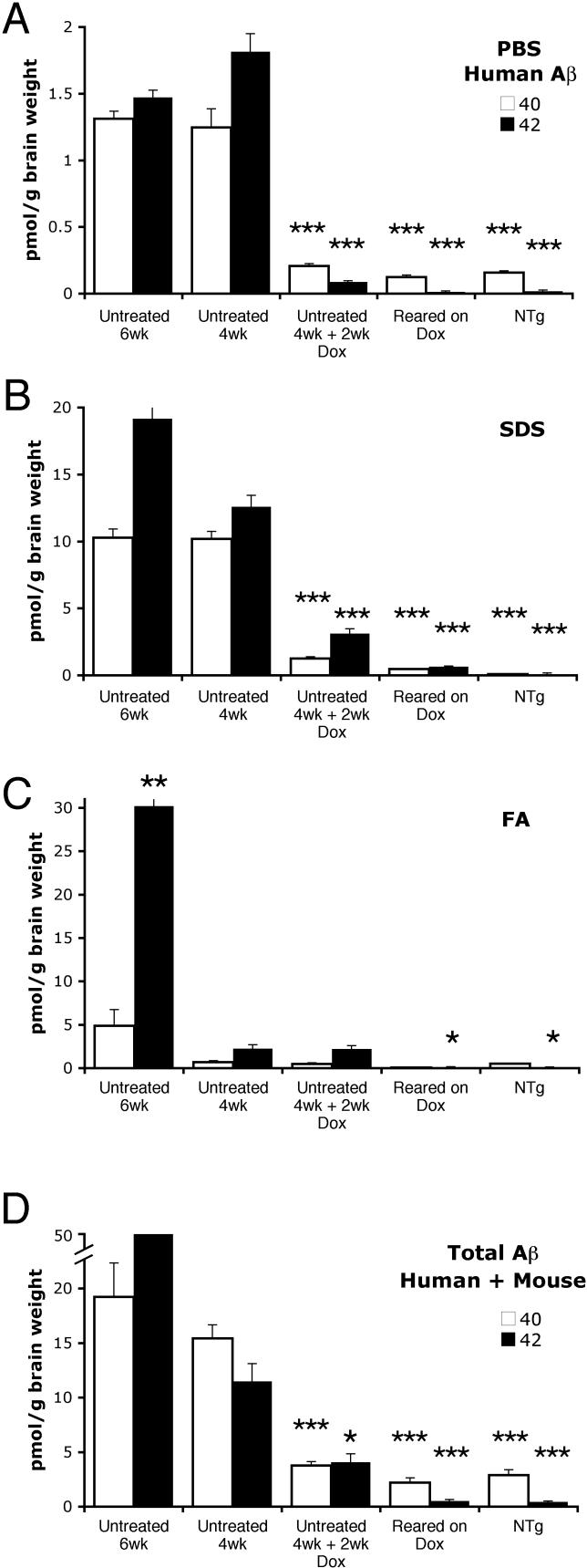
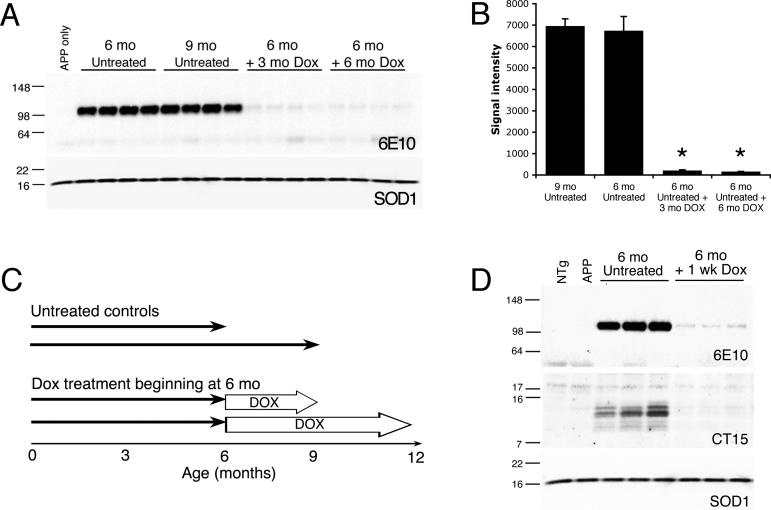
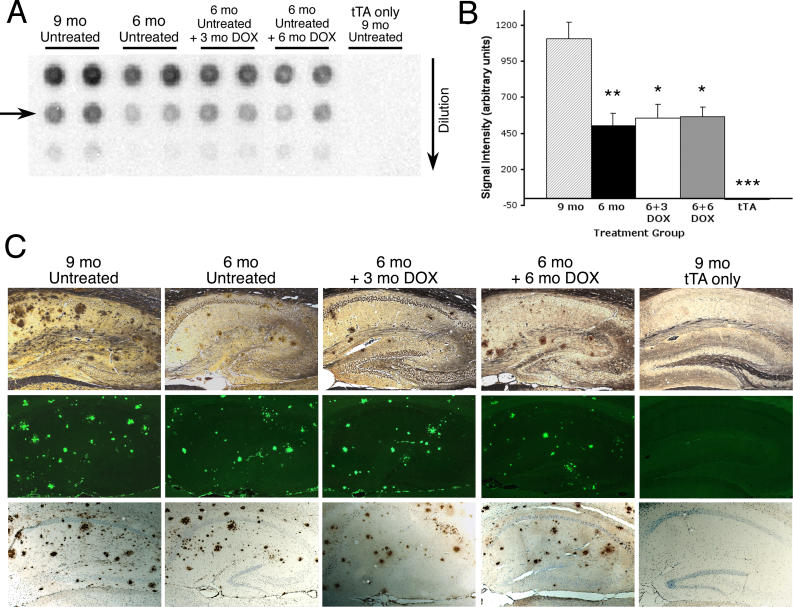
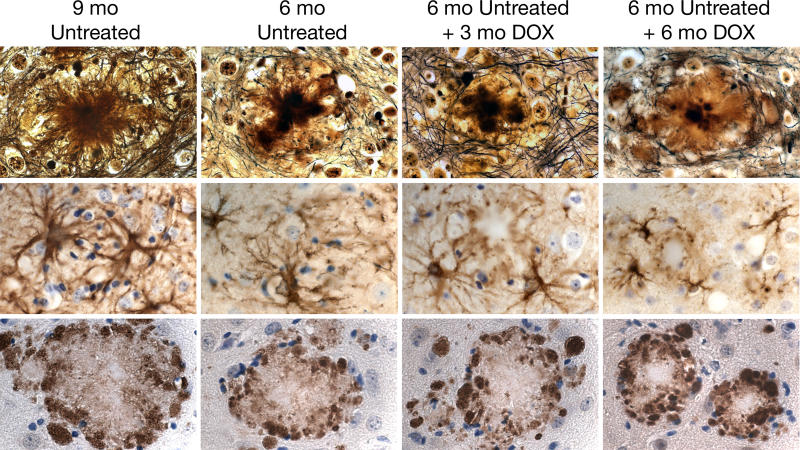
Methods and findings: We have generated a transgenic mouse model that genetically mimics the arrest of Abeta production expected from treatment with secretase inhibitors. These mice overexpress mutant APP from a vector that can be regulated by doxycycline. Under normal conditions, high-level expression of APP quickly induces fulminant amyloid pathology. We show that doxycycline administration inhibits transgenic APP expression by greater than 95% and reduces Abeta production to levels found in nontransgenic mice. Suppression of transgenic Abeta synthesis in this model abruptly halts the progression of amyloid pathology. However, formation and disaggregation of amyloid deposits appear to be in disequilibrium as the plaques require far longer to disperse than to assemble. Mice in which APP synthesis was suppressed for as long as 6 mo after the formation of Abeta deposits retain a considerable amyloid load, with little sign of active clearance.
Conclusion: This study demonstrates that amyloid lesions in transgenic mice are highly stable structures in vivo that are slow to disaggregate. Our findings suggest that arresting Abeta production in patients with Alzheimer disease should halt the progression of pathology, but that early treatment may be imperative, as it appears that amyloid deposits, once formed, will require additional intervention to clear.
Conflict of interest statement
Figures



Comment in
-
Breaking up (amyloid) is hard to do.PLoS Med. 2005 Dec;2(12):e417. doi: 10.1371/journal.pmed.0020417. Epub 2005 Dec 27. PLoS Med. 2005. PMID: 16363913 Free PMC article.
References
-
- Hardy JA, Higgins GA. Alzheimer's disease: The amyloid cascade hypothesis. Science. 1992;256:184–185. - PubMed
-
- Selkoe DJ, Podlisny MB. Deciphering the genetic basis of Alzheimer's disease. Annu Rev Genomics Hum Genet. 2002;3:67–99. - PubMed
-
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, et al. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology. 2003;61:46–54. - PubMed
-
- Check E. Nerve inflammation halts trial for Alzheimer's drug. Nature. 2002;415:462. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
