

Molecular mechanisms of Sonic hedgehog mutant effects in holoprosencephaly
- PMID: 16282375
- PMCID: PMC1282174
- DOI: 10.1073/pnas.0507848102
Molecular mechanisms of Sonic hedgehog mutant effects in holoprosencephaly
Abstract
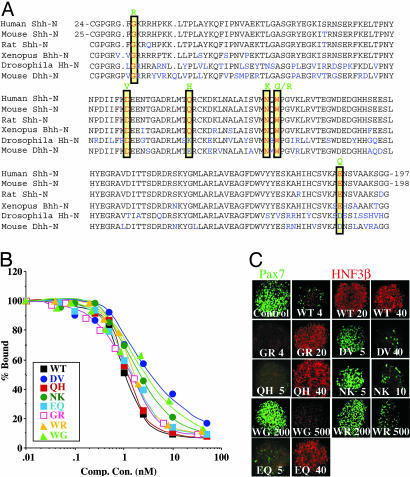
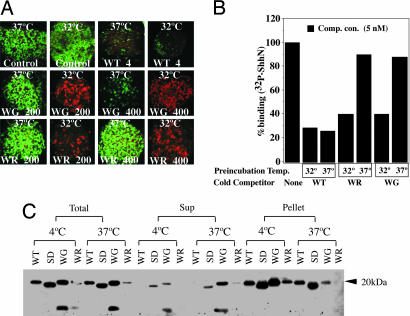
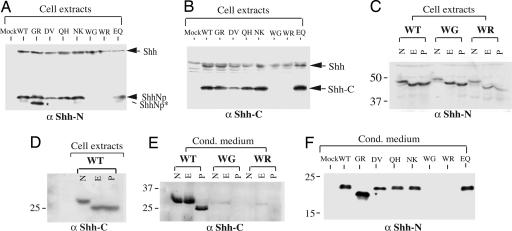
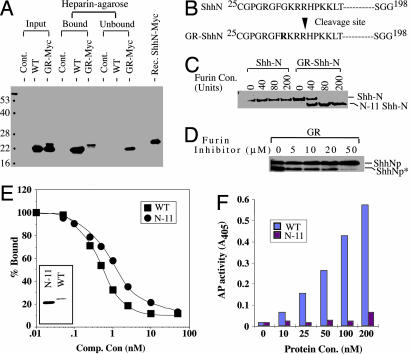
Holoprosencephaly (HPE), a human developmental brain defect, usually is also associated with varying degrees of midline facial dysmorphism. Heterozygous mutations in the Sonic hedgehog (SHH) gene are the most common genetic lesions associated with HPE, and loss of Shh function in the mouse produces cyclopia and alobar forebrain development. The N-terminal domain (ShhNp) of Sonic hedgehog protein, generated by cholesterol-dependent autoprocessing and modification at the C terminus and by palmitate addition at the N terminus, is the active ligand in the Shh signal transduction pathway. Here, we analyze seven reported missense mutations (G31R, D88V, Q100H, N115K, W117G, W117R, and E188Q) that alter the N-terminal signaling domain of Shh protein, and show that two of these mutations (Q100H and E188Q), which are questionably linked to HPE, produce no detectable effects on function. The remaining five alterations affect normal processing, Ptc binding, and signaling to varying degrees. These effects include introduction of a recognition site for furin-like proteases by the G31R alteration, resulting in cleavage of 11 amino acid residues from the N terminus of ShhNp and consequent reduced signaling potency. Two other alterations, W117G and W117R, cause temperature-dependent misfolding and retention in the sterol-poor endoplasmic reticulum, thus disrupting cholesterol-dependent autoprocessing.
Figures
Similar articles
-
Functional characterization of sonic hedgehog mutations associated with holoprosencephaly.J Biol Chem. 2004 Oct 8;279(41):42889-97. doi: 10.1074/jbc.M405161200. Epub 2004 Jul 28. J Biol Chem. 2004. PMID: 15292211
-
Mutations in the C-terminal domain of Sonic Hedgehog cause holoprosencephaly.Hum Mol Genet. 1997 Oct;6(11):1847-53. doi: 10.1093/hmg/6.11.1847. Hum Mol Genet. 1997. PMID: 9302262
-
Genetic approaches to understanding brain development: holoprosencephaly as a model.Ment Retard Dev Disabil Res Rev. 2000;6(1):15-21. doi: 10.1002/(SICI)1098-2779(2000)6:1<15::AID-MRDD3>3.0.CO;2-8. Ment Retard Dev Disabil Res Rev. 2000. PMID: 10899793 Review.
-
Ectopic sonic hedgehog signaling impairs telencephalic dorsal midline development: implication for human holoprosencephaly.Hum Mol Genet. 2007 Jun 15;16(12):1454-68. doi: 10.1093/hmg/ddm096. Epub 2007 Apr 27. Hum Mol Genet. 2007. PMID: 17468181
-
Mouse models of holoprosencephaly.Curr Opin Neurol. 2003 Apr;16(2):135-41. doi: 10.1097/01.wco.0000063761.15877.40. Curr Opin Neurol. 2003. PMID: 12644739 Review.
Cited by
-
Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth.Cancer Cell. 2010 Apr 13;17(4):388-99. doi: 10.1016/j.ccr.2010.02.027. Cancer Cell. 2010. PMID: 20385363 Free PMC article.
-
The molecular genetics of holoprosencephaly.Am J Med Genet C Semin Med Genet. 2010 Feb 15;154C(1):52-61. doi: 10.1002/ajmg.c.30236. Am J Med Genet C Semin Med Genet. 2010. PMID: 20104595 Free PMC article. Review.
-
Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases.Dis Model Mech. 2012 Nov;5(6):746-55. doi: 10.1242/dmm.010124. Epub 2012 Oct 12. Dis Model Mech. 2012. PMID: 23065638 Free PMC article. Review.
-
ABT-199 inhibits Hedgehog pathway by acting as a competitive inhibitor of oxysterol, rather as a BH3 mimetic.Acta Pharmacol Sin. 2021 Jun;42(6):1005-1013. doi: 10.1038/s41401-020-00504-4. Epub 2020 Aug 27. Acta Pharmacol Sin. 2021. PMID: 32855528 Free PMC article.
-
Zinc inhibits Hedgehog autoprocessing: linking zinc deficiency with Hedgehog activation.J Biol Chem. 2015 May 1;290(18):11591-600. doi: 10.1074/jbc.M114.623264. Epub 2015 Mar 18. J Biol Chem. 2015. PMID: 25787080 Free PMC article.
References
-
- Muenke, M. & Beachy, P. A. (2001) in The Metabolic and Molecular Bases of Inherited Disease, eds. Scriver, C., Beaudet, A., Sly, W. & Valle, D. (McGraw-Hill, New York), pp. 6203-6230.
-
- Chiang, C., Litingtung, Y., Lee, E., Young, K., Corden, J. L., Westphal, H. & Beachy, P. (1996) Nature 383, 407-413. - PubMed
-
- Ingham, P. W. & McMahon, A. P. (2001) Genes Dev. 15, 3059-3087. - PubMed
-
- Roessler, E., Belloni, E., Gaudenz, K., Jay, P., Berta, P., Scherer, S., Tsui, L. & Muenke, M. (1996) Nat. Genet. 14, 357-360. - PubMed
-
- Belloni, E., Muenke, M., Roessler, E., Traverso, G., Siegel-Bartelt, J., Frumkin, A., Mitchell, H., Donis-Keller, H., Helms, C., Hing, A., et al. (1996) Nat. Genet. 14, 353-356. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
