

Limitations and potentials of current motif discovery algorithms
- PMID: 16284194
- PMCID: PMC1199555
- DOI: 10.1093/nar/gki791
Limitations and potentials of current motif discovery algorithms
Abstract
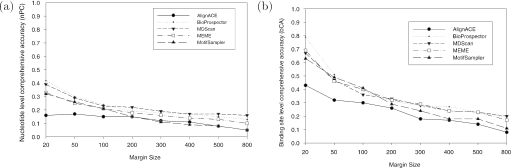
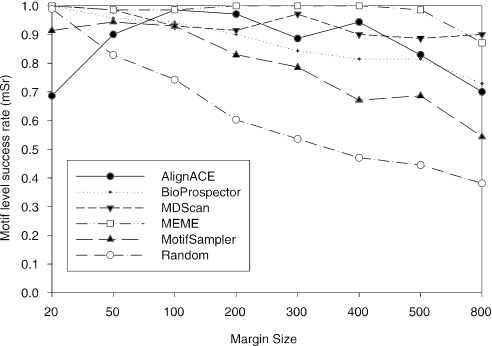
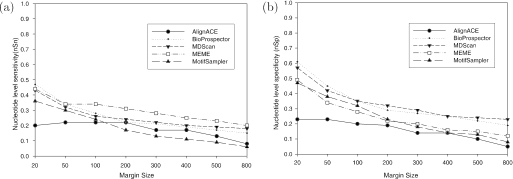
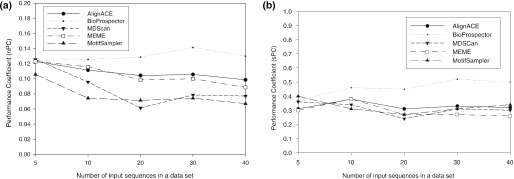
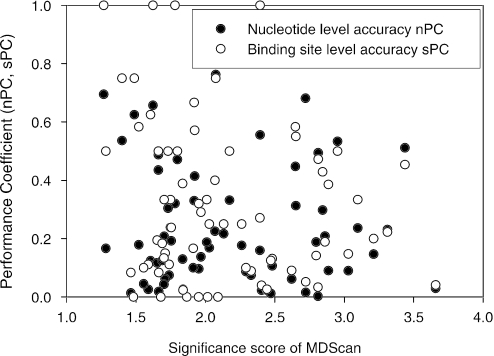
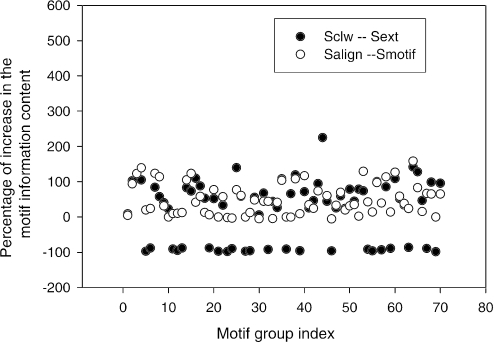
Computational methods for de novo identification of gene regulation elements, such as transcription factor binding sites, have proved to be useful for deciphering genetic regulatory networks. However, despite the availability of a large number of algorithms, their strengths and weaknesses are not sufficiently understood. Here, we designed a comprehensive set of performance measures and benchmarked five modern sequence-based motif discovery algorithms using large datasets generated from Escherichia coli RegulonDB. Factors that affect the prediction accuracy, scalability and reliability are characterized. It is revealed that the nucleotide and the binding site level accuracy are very low, while the motif level accuracy is relatively high, which indicates that the algorithms can usually capture at least one correct motif in an input sequence. To exploit diverse predictions from multiple runs of one or more algorithms, a consensus ensemble algorithm has been developed, which achieved 6-45% improvement over the base algorithms by increasing both the sensitivity and specificity. Our study illustrates limitations and potentials of existing sequence-based motif discovery algorithms. Taking advantage of the revealed potentials, several promising directions for further improvements are discussed. Since the sequence-based algorithms are the baseline of most of the modern motif discovery algorithms, this paper suggests substantial improvements would be possible for them.
Figures
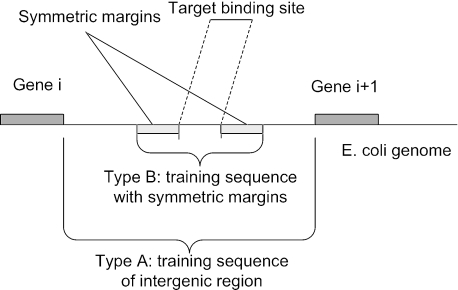
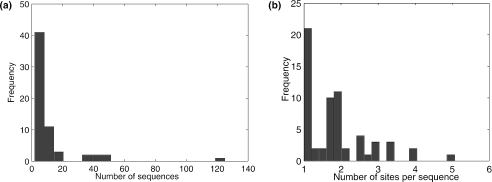
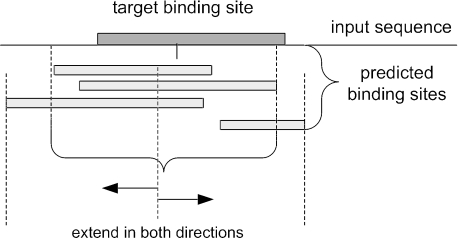
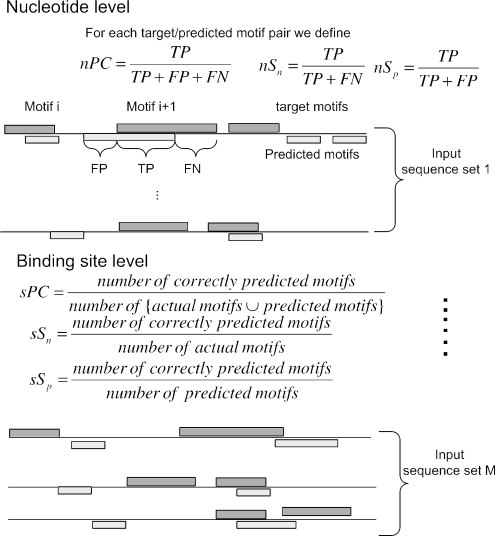

References
-
- Wyrick J.J., Young R.A. Deciphering gene expression regulatory networks. Curr. Opin. Genet. Dev. 2002;12:130–136. - PubMed
-
- Duret L., Bucher P. Searching for regulatory elements in human noncoding sequences. Curr. Opin. Struct. Biol. 1997;7:399–406. - PubMed
-
- Simon I., Barnett J., Hannett N., Harbison C.T., Rinaldi N.J., Volkert T.L., Wyrick J.J., Zeitlinger J., Gifford D.K., Jaakkola T.S., Young R.A. Serial regulation of transcriptional regulators in the yeast cell cycle. Cell. 2001;106:697–708. - PubMed
