

The mitochondrial origin of postischemic arrhythmias
- PMID: 16284648
- PMCID: PMC1280968
- DOI: 10.1172/JCI25371
The mitochondrial origin of postischemic arrhythmias
Abstract
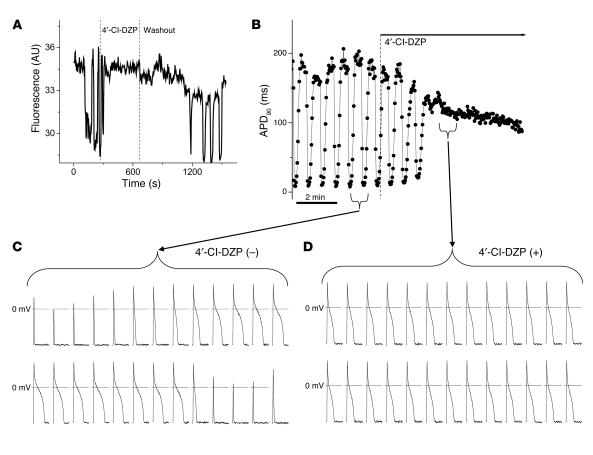
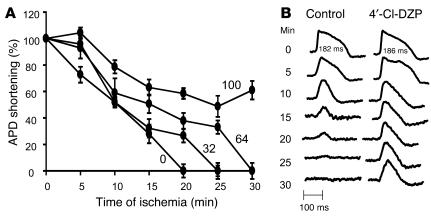
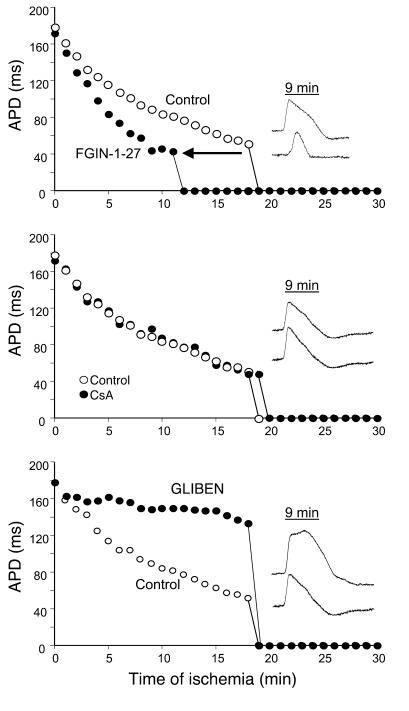
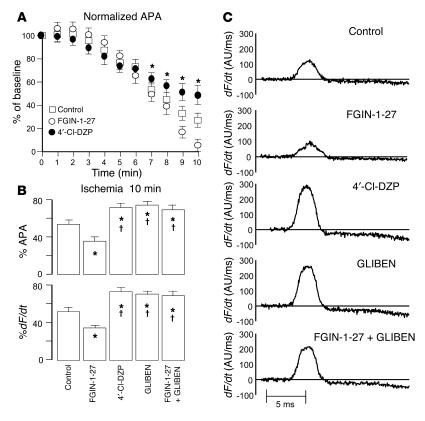
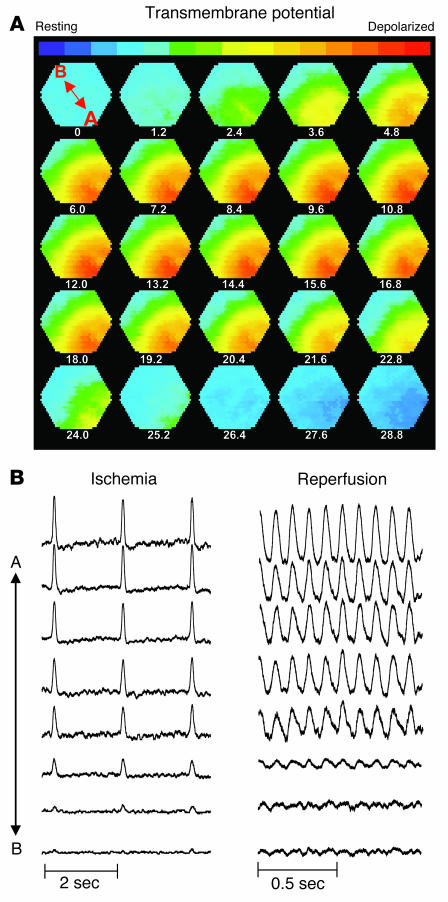
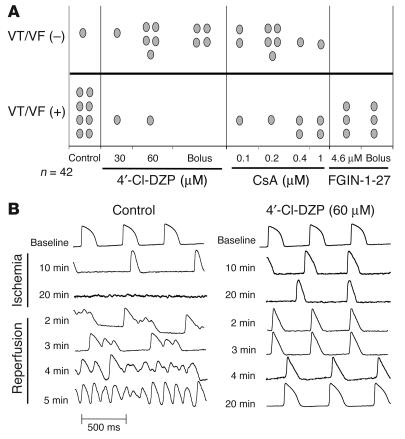
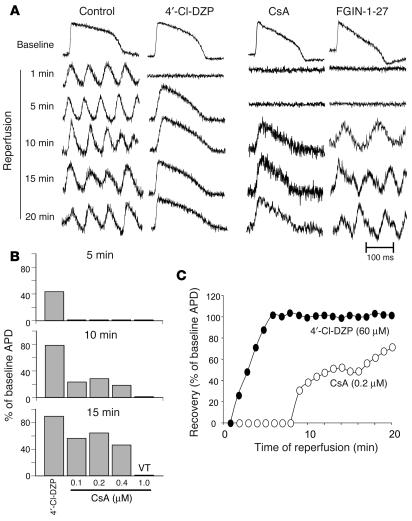
Recovery of the mitochondrial inner membrane potential (DeltaPsi(m)) is a key determinant of postischemic functional recovery of the heart. Mitochondrial ROS-induced ROS release causes the collapse of DeltaPsi(m) and the destabilization of the action potential (AP) through a mechanism involving a mitochondrial inner membrane anion channel (IMAC) modulated by the mitochondrial benzodiazepine receptor (mBzR). Here, we test the hypothesis that this mechanism contributes to spatiotemporal heterogeneity of DeltaPsi(m) during ischemia-reperfusion (IR), thereby promoting abnormal electrical activation and arrhythmias in the whole heart. High-resolution optical AP mapping was performed in perfused guinea pig hearts subjected to 30 minutes of global ischemia followed by reperfusion. Typical electrophysiological responses, including progressive AP shortening followed by membrane inexcitablity in ischemia and ventricular fibrillation upon reperfusion, were observed in control hearts. These responses were reduced or eliminated by treatment with the mBzR antagonist 4'-chlorodiazepam (4'-Cl-DZP), which blocks depolarization of DeltaPsi(m). When applied throughout the IR protocol, 4'-Cl-DZP blunted AP shortening and prevented reperfusion arrhythmias. Inhibition of ventricular fibrillation was also achieved by bolus infusion of 4'-Cl-DZP just before reperfusion. Conversely, treatment with an agonist of the mBzR that promotes DeltaPsi(m) depolarization exacerbated IR-induced electrophysiological changes and failed to prevent arrhythmias. The effects of these compounds were consistent with their actions on IMAC and DeltaPsi(m). These findings directly link instability of DeltaPsi(m) to the heterogeneous electrophysiological substrate of the postischemic heart and highlight the mitochondrial membrane as a new therapeutic target for arrhythmia prevention in ischemic heart disease.
Figures
References
-
- Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol. Rev. 1999;79:917–1017. - PubMed
-
- Coronel R, et al. Reperfusion arrhythmias in isolated perfused pig hearts. Inhomogeneities in extracellular potassium, ST and TQ potentials, and transmembrane action potentials. Circ. Res. 1992;71:1131–1142. - PubMed
-
- De Groot JR, Coronel R. Acute ischemia-induced gap junctional uncoupling and arrhythmogenesis. Cardiovasc. Res. 2004;62:323–334. - PubMed
-
- Crompton M, Virji S, Doyle V, Johnson N, Ward JM. The mitochondrial permeability transition pore. Biochem. Soc. Symp. 1999;66:167–179. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
