

Critical role of calcitonin gene-related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior
- PMID: 16291945
- PMCID: PMC6725858
- DOI: 10.1523/JNEUROSCI.4112-05.2005
Critical role of calcitonin gene-related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior
Abstract
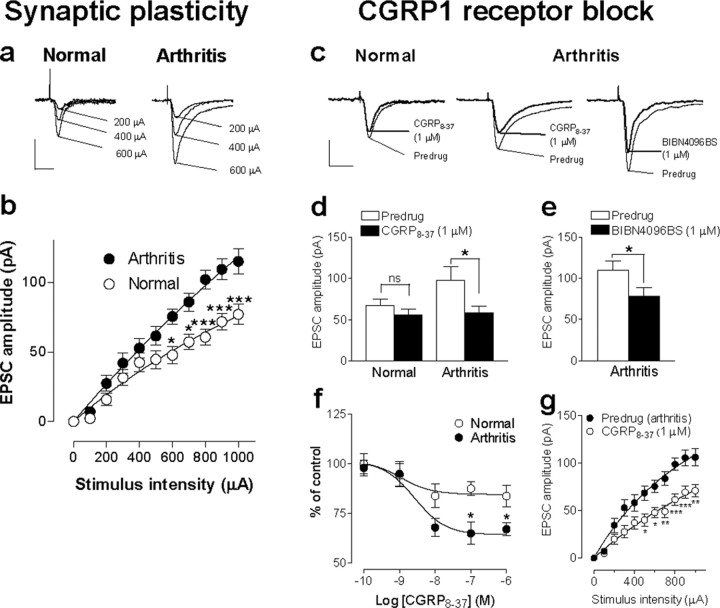
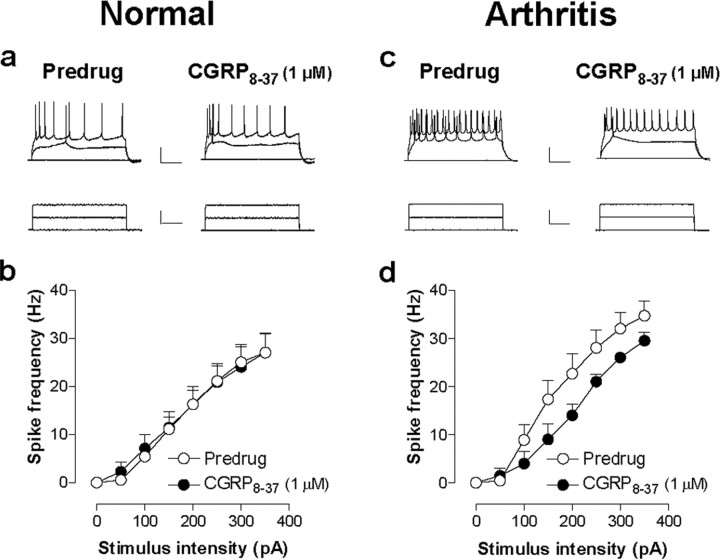
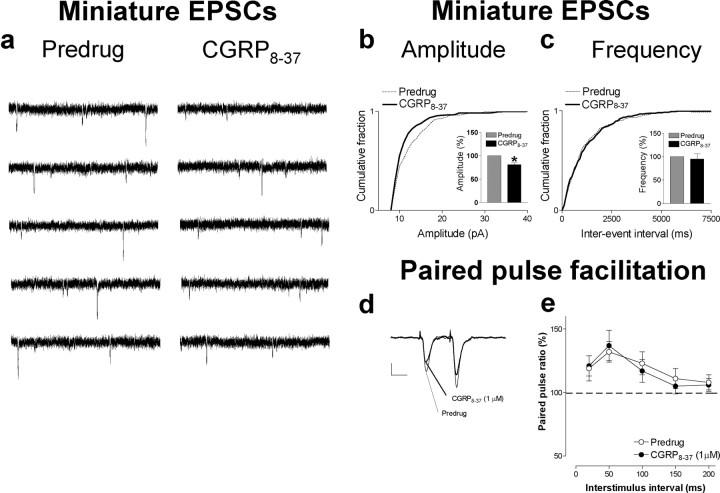
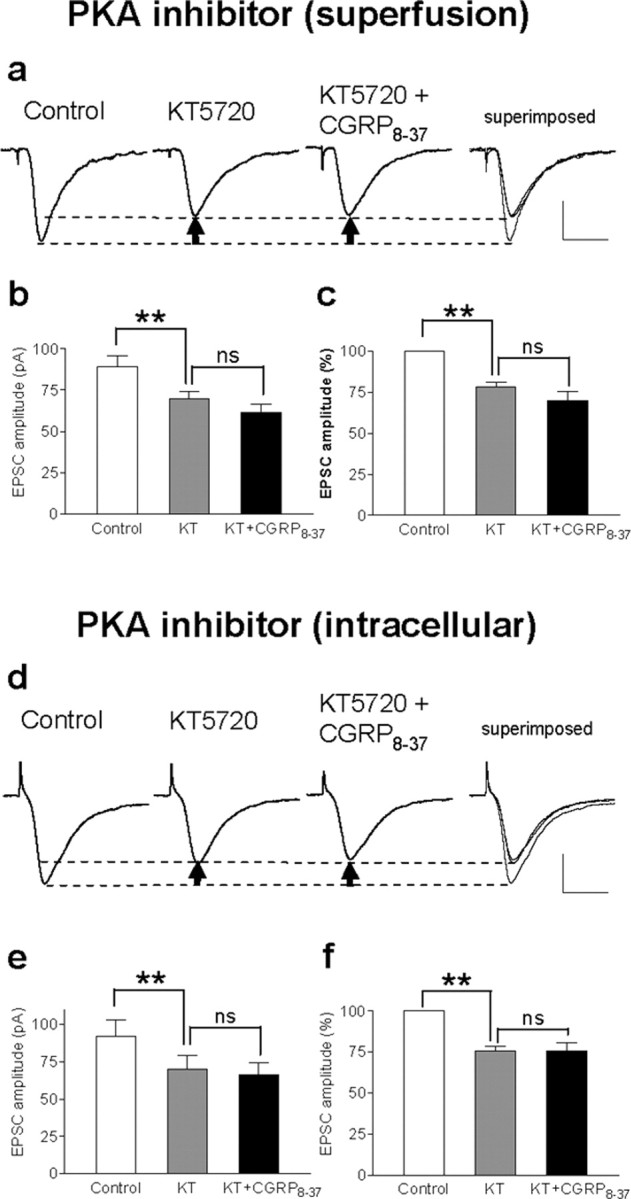
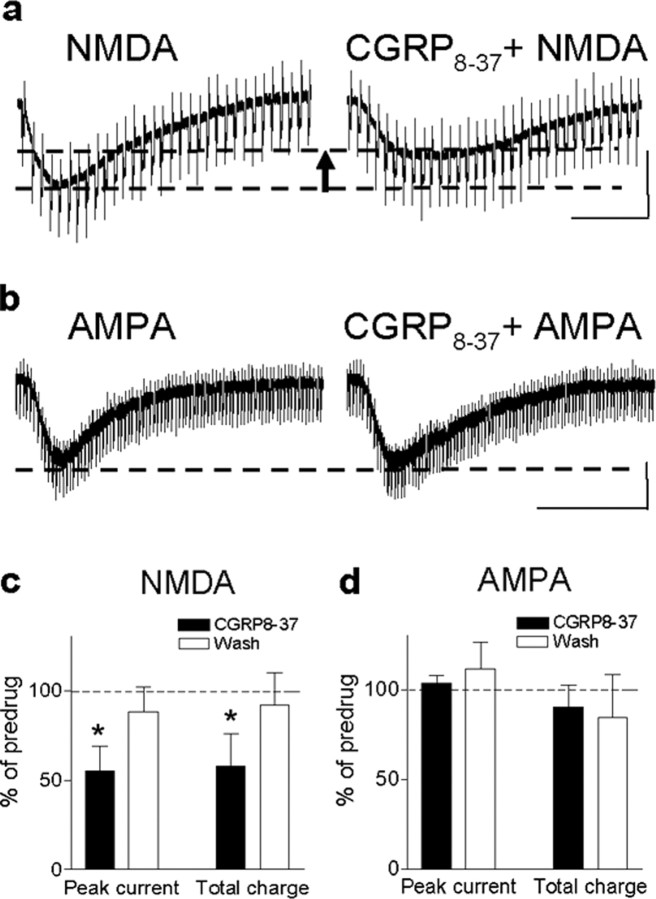
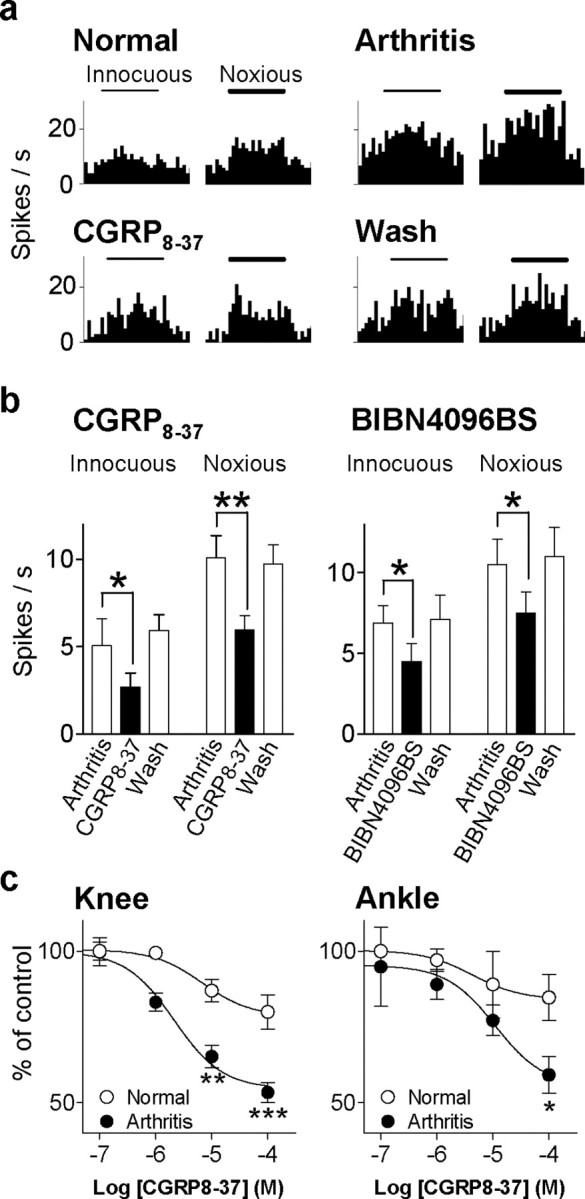
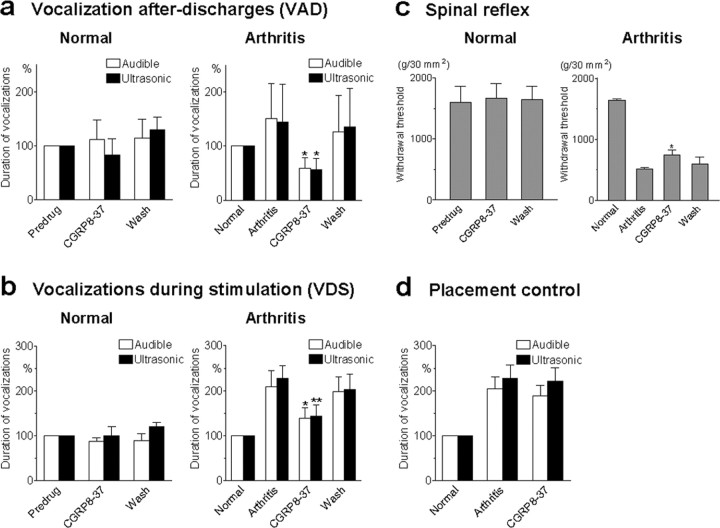
The role of neuropeptides in synaptic plasticity is less well understood than that of classical transmitters such as glutamate. Here we report the importance of the G-protein-coupled calcitonin gene-related peptide (CGRP1) receptor as a critical link between amygdala plasticity and pain behavior. A key player in emotionality and affective disorders, the amygdala has been implicated in the well documented, but mechanistically unexplained, relationship between pain and affect. Our electrophysiological and pharmacological in vitro (patch-clamp recordings) and in vivo (extracellular single-unit recordings) data show that selective CGRP1 receptor antagonists (CGRP(8-37) and BIBN4096BS) in the amygdala reverse arthritis pain-related plasticity through a protein kinase A (PKA)-dependent postsynaptic mechanism that involves NMDA receptors. CGRP1 receptor antagonists inhibited synaptic plasticity in the laterocapsular division of the central nucleus of the amygdala (CeLC) in brain slices from arthritic rats compared with normal controls. The effects were accompanied by decreased neuronal excitability and reduced amplitude, but not frequency, of miniature EPSCs; paired-pulse facilitation was unaffected. The antagonist effects were occluded by a PKA inhibitor. CGRP1 receptor blockade also directly inhibited NMDA-evoked, but not AMPA-evoked, membrane currents. Together, these data suggest a postsynaptic site of action. At the systems level, the antagonists reversed the sensitization of nociceptive CeLC neurons in anesthetized rats in the arthritis pain model. Importantly, CGRP1 receptor blockade in the CeLC inhibited spinal (hindlimb withdrawal reflexes) and supraspinal pain behavior of awake arthritic rats, including affective responses such as ultrasonic vocalizations. This study provides direct evidence for the critical dependence of pain behavior on CGRP1-mediated amygdala plasticity.
Figures
References
-
- Bernard J-F, Bandler R (1998) Parallel circuits for emotional coping behaviour: new pieces in the puzzle. J Comp Neurol 401: 429-436. - PubMed
-
- Borszcz GS, Leaton RN (2003) The effect of amygdala lesions on conditional and unconditional vocalizations in rats. Neurobiol Learn Mem 79: 212-225. - PubMed
-
- Cabell L, Audesirk G (1993) Effects of selective inhibition of protein kinase C, cyclic AMP-dependent protein kinase, and Ca2+-calmodulin-dependent protein kinase on neurite development in cultured rat hippocampal neurons. Int J Dev Neurosci 11: 357-368. - PubMed
-
- Cartmell J, Schoepp DD (2000) Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem 75: 889-907. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials