

Evidence of a large novel gene pool associated with prokaryotic genomic islands
- PMID: 16299586
- PMCID: PMC1285063
- DOI: 10.1371/journal.pgen.0010062
Evidence of a large novel gene pool associated with prokaryotic genomic islands
Abstract
Microbial genes that are "novel" (no detectable homologs in other species) have become of increasing interest as environmental sampling suggests that there are many more such novel genes in yet-to-be-cultured microorganisms. By analyzing known microbial genomic islands and prophages, we developed criteria for systematic identification of putative genomic islands (clusters of genes of probable horizontal origin in a prokaryotic genome) in 63 prokaryotic genomes, and then characterized the distribution of novel genes and other features. All but a few of the genomes examined contained significantly higher proportions of novel genes in their predicted genomic islands compared with the rest of their genome (Paired t test = 4.43E-14 to 1.27E-18, depending on method). Moreover, the reverse observation (i.e., higher proportions of novel genes outside of islands) never reached statistical significance in any organism examined. We show that this higher proportion of novel genes in predicted genomic islands is not due to less accurate gene prediction in genomic island regions, but likely reflects a genuine increase in novel genes in these regions for both bacteria and archaea. This represents the first comprehensive analysis of novel genes in prokaryotic genomic islands and provides clues regarding the origin of novel genes. Our collective results imply that there are different gene pools associated with recently horizontally transmitted genomic regions versus regions that are primarily vertically inherited. Moreover, there are more novel genes within the gene pool associated with genomic islands. Since genomic islands are frequently associated with a particular microbial adaptation, such as antibiotic resistance, pathogen virulence, or metal resistance, this suggests that microbes may have access to a larger "arsenal" of novel genes for adaptation than previously thought.
Conflict of interest statement
Competing interests. The authors have declared that no competing interests exist.
Figures
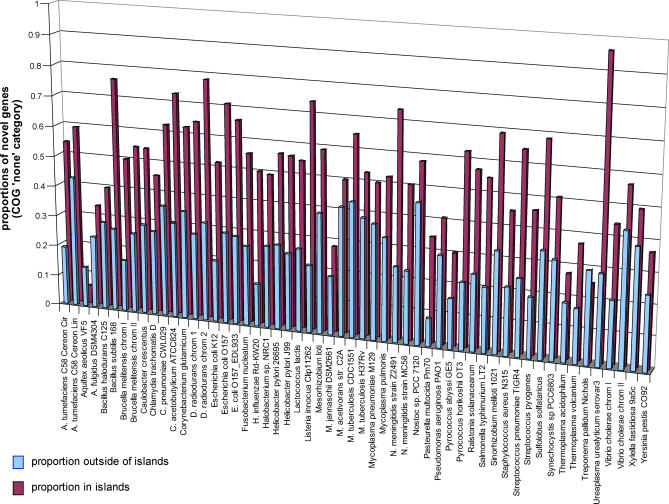
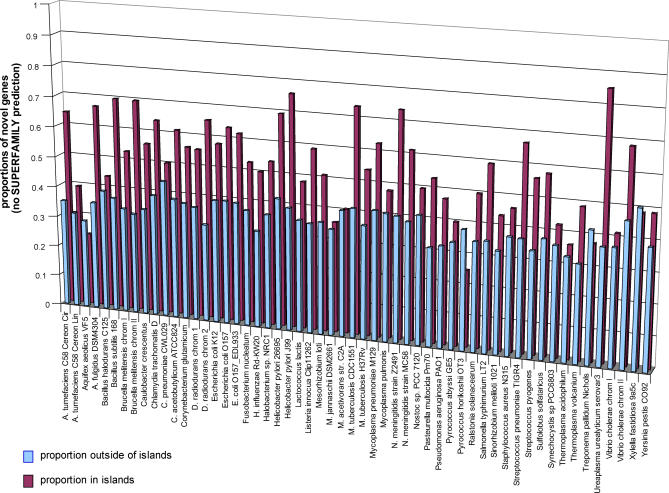
References
-
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. - PubMed
-
- Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis . Nature. 1997;390:249–256. - PubMed
-
- Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. - PubMed
-
- Bork P. Powers and pitfalls in sequence analysis: The 70% hurdle. Genome Res. 2000;10:398–400. - PubMed
-
- Siew N, Fischer D. Analysis of singleton ORFans in fully sequenced microbial genomes. Proteins. 2003;53:241–251. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
