

Recombination in the genesis and evolution of hepatitis B virus genotypes
- PMID: 16306618
- PMCID: PMC1316029
- DOI: 10.1128/JVI.79.24.15467-15476.2005
Recombination in the genesis and evolution of hepatitis B virus genotypes
Abstract
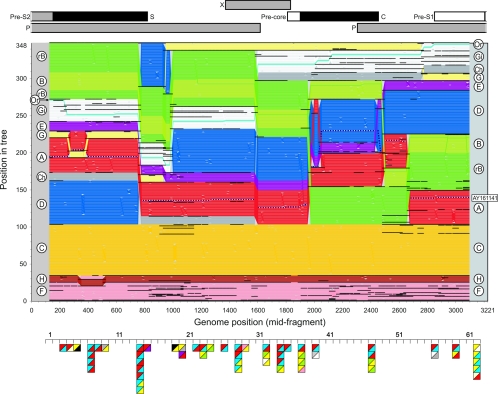
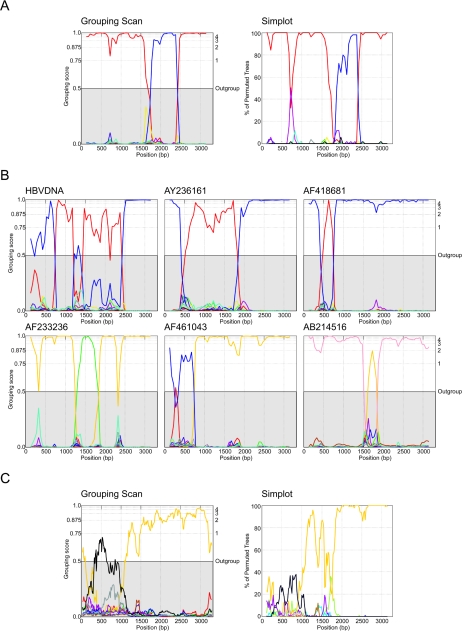
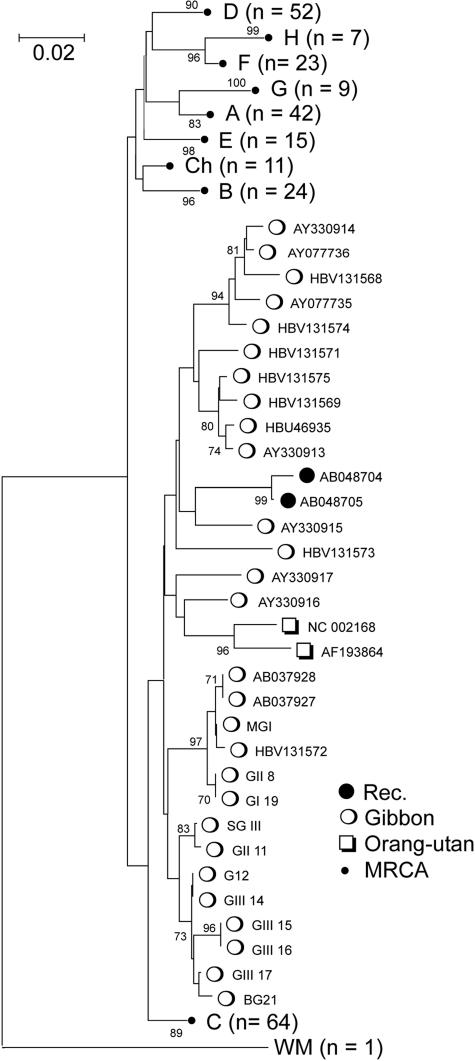
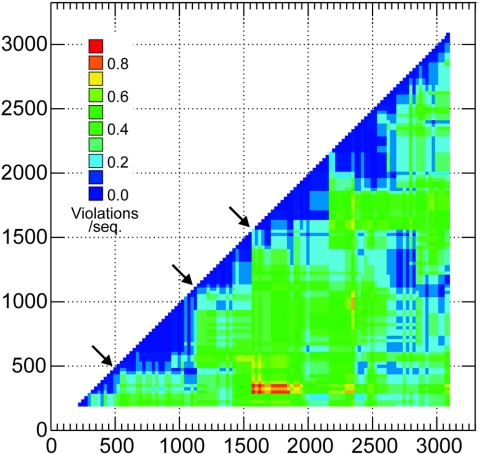
Hepatitis B virus (HBV) infection is widely distributed in both human and ape populations throughout the world and is a major cause of human morbidity and mortality. HBV variants are currently classified into the human genotypes A to H and species-associated chimpanzee and gibbon/orangutan groups. To examine the role of recombination in the evolution of HBV, large-scale data retrieval and automated phylogenetic analysis (TreeOrder scanning) were carried out on all available published complete genome sequences of HBV. We detected a total of 24 phylogenetically independent potential recombinants (different genotype combinations or distinct breakpoints), eight of which were previously undescribed. Instances of intergenotype recombination were observed in all human and ape HBV variants, including evidence for a novel gibbon/genotype C recombinant among HBV variants from Vietnam. By recording sequence positions in trees generated from sequential fragments across the genome, violations of phylogeny between trees also provided evidence for frequent intragenotype recombination between members of genotypes A, D, F/H, and gibbon variants but not in B, C, or the Asian B/C recombinant group. In many cases, favored positions for both inter- and intragenotype recombination matched positions of phylogenetic reorganization between the human and ape genotypes, such as the end of the surface gene and the core gene, where sequence relationships between genotypes changed in the TreeOrder scan. These findings provide evidence for the occurrence of past, extensive recombination events in the evolutionary history of the currently classified genotypes of HBV and potentially in changes in its global epidemiology and associations with human disease.
Figures
References
-
- Akuta, N., and H. Kumada. 2005. Influence of hepatitis B virus genotypes on the response to antiviral therapies. J. Antimicrob. Chemother. 55:139-142. - PubMed
-
- Andre, F. 2000. Hepatitis B epidemiology in Asia, the Middle East and Africa. Vaccine 18(Suppl. 1):S20-S22. - PubMed
-
- Bollyky, P. L., and E. C. Holmes. 1999. Reconstructing the complex evolutionary history of hepatitis B virus. J. Mol. Evol. 49:130-141. - PubMed
-
- Bollyky, P. L., A. Rambaut, P. H. Harvey, and E. C. Holmes. 1996. Recombination between sequences of hepatitis B virus from different genotypes. J. Mol. Evol. 42:97-102. - PubMed
-
- Bowyer, S. M., and J. G. Sim. 2000. Relationships within and between genotypes of hepatitis B virus at points across the genome: footprints of recombination in certain isolates. J. Gen. Virol. 81:379-392. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
